
Identification and Characterization of a Novel Recurrent ERCC6 Variant in Patients with a Severe Form of Cockayne Syndrome B


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. DNA Extraction, Quantification, and Quality Control
2.3. gDNA Sequencing
2.3.1. Targeted Next Generation Sequencing
2.3.2. Targeted Sanger Sequencing
2.4. DNA Repair Assay in Primary Dermal Fibroblasts
2.5. Western Blotting
3. Results
3.1. Clinical Features of CS-B Patients
3.1.1. General Presentation of the Patients
3.1.2. Pre- and Post-Natal Abnormalities
3.1.3. Behavioral Abnormalities, and Muscular, Neurological, and Neurosensory Problems
3.1.4. Facial, Dental, and Skin Anomalies
3.1.5. Laboratory Investigations
3.1.6. Neuroimaging Analysis
3.1.7. Neurophysiological Studies
3.2. Genetic and Biochemical Analyses
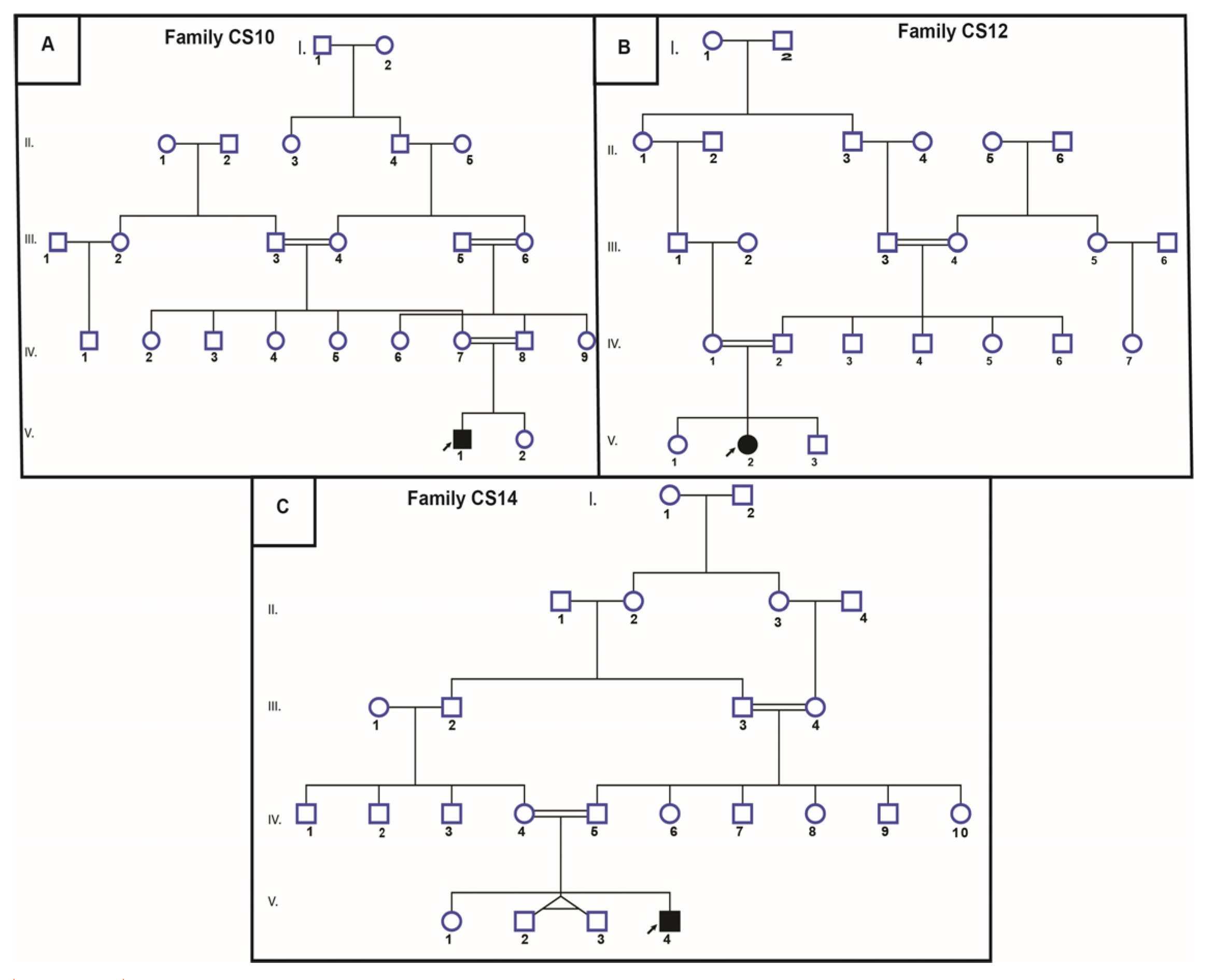
3.2.1. Sanger Sequencing for Recurrent Mutations
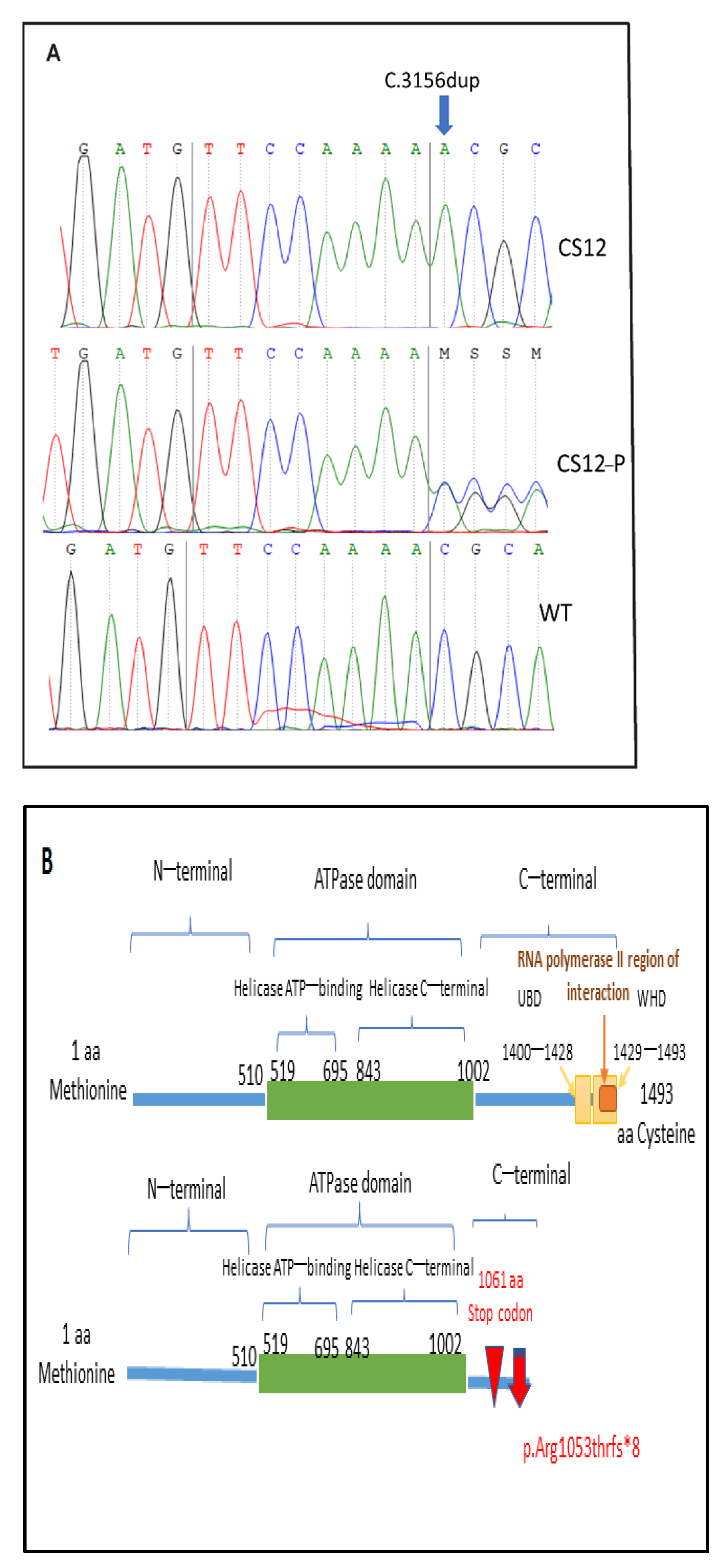
3.2.2. Targeted Gene Sequencing Finding and Sanger Sequencing Validation for the Novel Variant
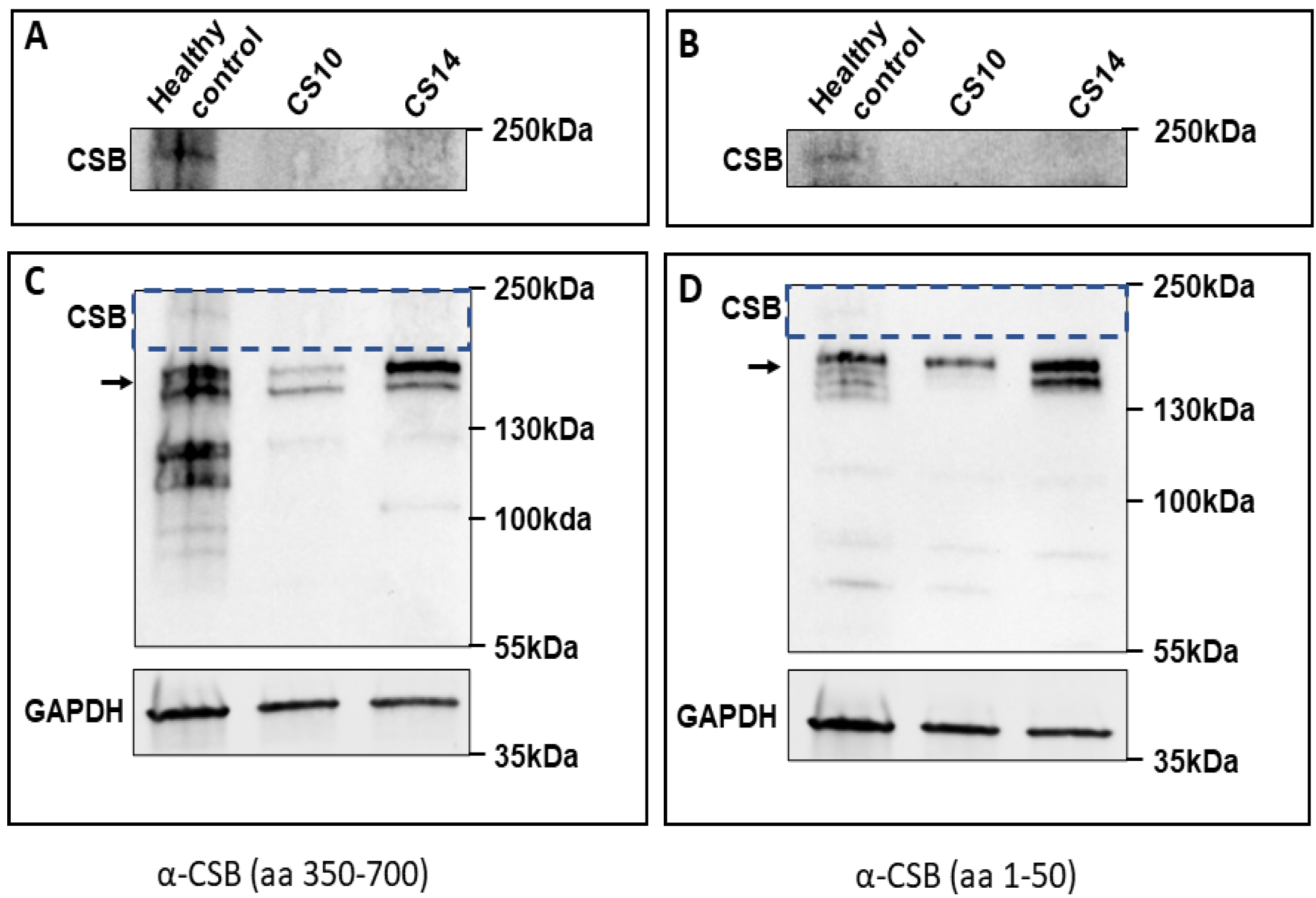
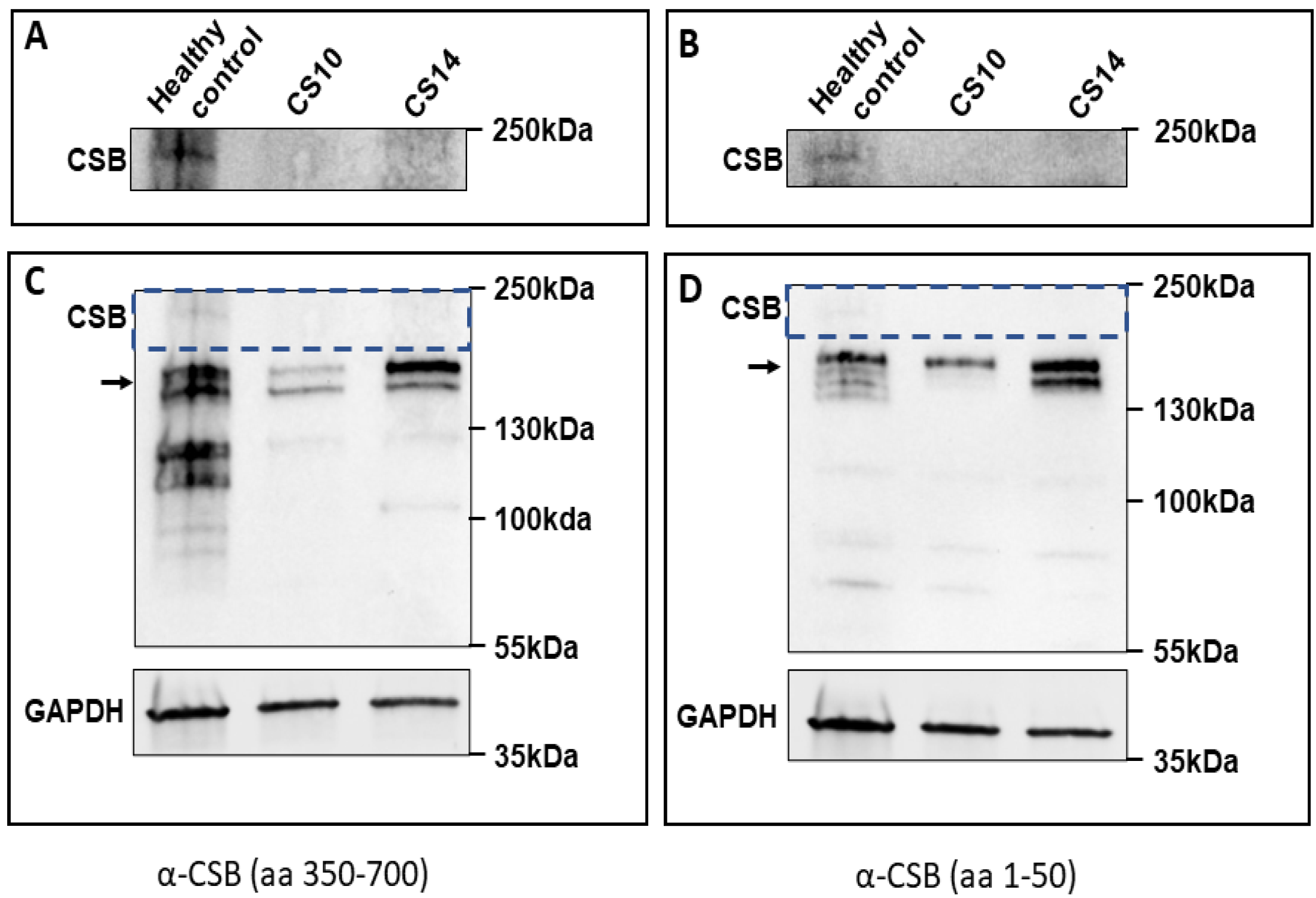
3.3. Western Blot Validation of Protein Alteration
3.4. Cellular Response to UV in CS Patients
4. Discussion
4.1. Common Mutation in Three CS Patients
4.2. The Mutated CSB Protein
4.3. Remarkable Clinical Features and Presence or Absence of Clinical Photosensitivity
4.4. Severe Characteristics of the Tunisian CSB Cohort
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Calmels, N.; Botta, E.; Jia, N.; Fawcett, H.; Nardo, T.; Nakazawa, Y.; Lanzafame, M.; Moriwaki, S.; Sugita, K.; Kubota, M.; et al. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 2018, 55, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.T.; Stark, Z.; Sutton, R.E.; Danda, S.; Ekbote, A.V.; Elsayed, S.M.; Gibson, L.; Goodship, J.A.; Jackson, A.P.; Keng, W.T.; et al. The Cockayne Syndrome Natural History (CoSyNH) study: Clinical findings in 102 individuals and recommendations for care. Genet. Med. 2016, 18, 483–493. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. Mitochondrial deficiency in Cockayne syndrome. Mech. Ageing Dev. 2013, 134, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, A.; Asghar, M.; Ryan, S.; Lynch, B.; Green, A.; Knerr, I. GP59 A rare cause of ‘mitochondrial disorder’: Cockayne syndrome. Arch. Dis. Child. 2019, 104, A53. [Google Scholar] [CrossRef] [Green Version]
- Laugel, V. Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 2013, 134, 161–170. [Google Scholar] [CrossRef]
- Narayanan, D.L.; Tuteja, M.; McIntyre, A.D.; Hegele, R.A.; Calmels, N.; Obringer, C.; Laugel, V.; Mandal, K.; Phadke, S.R. Clinical and Mutation Spectra of Cockayne Syndrome in India. Neurol. India 2021, 69, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kciuk, M.; Marciniak, B.; Mojzych, M.; Kontek, R. Focus on UV-Induced DNA Damage and Repair-Disease Relevance and Protective Strategies. Int. J. Mol. Sci. 2020, 21, 7264. [Google Scholar] [CrossRef]
- Pines, A.; Dijk, M.; Makowski, M.; Meulenbroek, E.M.; Vrouwe, M.G.; van der Weegen, Y.; Baltissen, M.; French, P.J.; van Royen, M.E.; Luijsterburg, M.S.; et al. TRiC controls transcription resumption after UV damage by regulating Cockayne syndrome protein A. Nat. Commun. 2018, 9, 1040. [Google Scholar] [CrossRef]
- Zghal, M.; El-Fekih, N.; Fazaa, B.; Fredj, M.; Zhioua, R.; Mokhtar, I.; Mrabet, A.; Ferjani, M.; Gaigi, S.; Kamoun, M.R. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 49 Tunisian cases. La Tunis. Med. 2005, 83, 760–763. [Google Scholar]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef]
- Natale, V. A comprehensive description of the severity groups in Cockayne syndrome. Am. J. Med. Genet. Part A 2011, 155a, 1081–1095. [Google Scholar] [CrossRef]
- Bloch-Zupan, A.; Rousseaux, M.; Laugel, V.; Schmittbuhl, M.; Mathis, R.; Desforges, E.; Koob, M.; Zaloszyc, A.; Dollfus, H.; Laugel, V. A possible cranio-oro-facial phenotype in Cockayne syndrome. Orphanet J. Rare Dis. 2013, 8, 9. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome group B (CSB) protein. Nucleic Acids Res. 2021, 49, 2418–2434. [Google Scholar] [CrossRef]
- Kou, Y.; Shboul, M.; Wang, Z.; Shersheer, Q.; Lyu, Z.; Liu, P.; Zhao, X.; Tian, J. Novel frame shift mutation in ERCC6 leads to a severe form of Cockayne syndrome with postnatal growth failure and early death: A case report and brief literature review. Medicine 2018, 97, e11636. [Google Scholar] [CrossRef]
- Tinsa, F.; Bellalah, M.; Brini, I.; Bousnina, D.; Lehmann, A.; Boussetta, K.; Bousnina, S. Infantile onset of Cockayne syndrome without photosensitivity in a Tunisian girl. La Tunis. Med. 2009, 87, 877–879. [Google Scholar]
- Vessoni, A.T.; Guerra, C.C.C.; Kajitani, G.S.; Nascimento, L.L.S.; Garcia, C.C.M. Cockayne Syndrome: The many challenges and approaches to understand a multifaceted disease. Genet. Mol. Biol. 2020, 43, e20190085. [Google Scholar] [CrossRef] [PubMed]
- Calmels, N.; Greff, G.; Obringer, C.; Kempf, N.; Gasnier, C.; Tarabeux, J.; Miguet, M.; Baujat, G.; Bessis, D.; Bretones, P.; et al. Uncommon nucleotide excision repair phenotypes revealed by targeted high-throughput sequencing. Orphanet J. Rare Dis. 2016, 11, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khayat, M.; Hardouf, H.; Zlotogora, J.; Shalev, S.A. High carriers frequency of an apparently ancient founder mutation p.Tyr322X in the ERCC8 gene responsible for Cockayne syndrome among Christian Arabs in Northern Israel. Am. J. Med. Genet. Part A 2010, 152a, 3091–3094. [Google Scholar] [CrossRef] [PubMed]
- Ben Chehida, A.; Ghali, N.; Ben Abdelaziz, R.; Ben Moussa, F.; Tebib, N. Renal Involvement in 2 Siblings With Cockayne Syndrome. Iran. J. Kidney Dis. 2017, 11, 253–255. [Google Scholar]
- Blin-Rochemaure, N.; Allani-Essid, N.; Carlier, R.; Laugel, V.; Quijano-Roy, S. Place de la neuropathie dans le diagnostic précoce du syndrome de Cockayne : À propos de deux cas dans une fratrie. Arch. Pédiatrie 2017, 24, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kaissi, A.; Safi, H.; Ghachem, M.; Hendaoui, L.; Chehida, F. Congenital dysplastic hips, spinal column abnormalities, fractures and progressive neurological manifestations in Tunisian family with cockayne syndrome. Ann. Afr. Med. 2005, 4, 83–87. [Google Scholar]
- Geoffroy, V.; Pizot, C.; Redin, C.; Piton, A.; Vasli, N.; Stoetzel, C.; Blavier, A.; Laporte, J.; Muller, J. VaRank: A simple and powerful tool for ranking genetic variants. PeerJ 2015, 3, e796. [Google Scholar] [CrossRef] [PubMed]
- Backenroth, D.; Homsy, J.; Murillo, L.R.; Glessner, J.; Lin, E.; Brueckner, M.; Lifton, R.; Goldmuntz, E.; Chung, W.K.; Shen, Y. CANOES: Detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 2014, 42, e97. [Google Scholar] [CrossRef]
- Geoffroy, V.; Herenger, Y.; Kress, A.; Stoetzel, C.; Piton, A.; Dollfus, H.; Muller, J. AnnotSV: An integrated tool for structural variations annotation. Bioinformatics 2018, 34, 3572–3574. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Yamashita, S.; Lehmann, A.R.; Ogi, T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair 2010, 9, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Jia, N.; Nakazawa, Y.; Guo, C.; Shimada, M.; Sethi, M.; Takahashi, Y.; Ueda, H.; Nagayama, Y.; Ogi, T. A rapid, comprehensive system for assaying DNA repair activity and cytotoxic effects of DNA-damaging reagents. Nat. Protoc. 2015, 10, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Calmels, N.; Abiayad, Y.; Boukli, L.; Semer, M.; Serradj, A.; Egly, J.-M.; Laugel, L. Xeroderma pigmentosum groups C and A in Algerian patients with deregulation of both transcription and DNA repair. J. Case Rep. Stud. 2018, 6. [Google Scholar] [CrossRef]
- Gitiaux, C.; Blin-Rochemaure, N.; Hully, M.; Echaniz-Laguna, A.; Calmels, N.; Bahi-Buisson, N.; Desguerre, I.; Dabaj, I.; Wehbi, S.; Quijano-Roy, S.; et al. Progressive demyelinating neuropathy correlates with clinical severity in Cockayne syndrome. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2015, 126, 1435–1439. [Google Scholar] [CrossRef]
- Newman, J.C.; Bailey, A.D.; Fan, H.Y.; Pavelitz, T.; Weiner, A.M. An abundant evolutionarily conserved CSB-PiggyBac fusion protein expressed in Cockayne syndrome. PLoS Genet. 2008, 4, e1000031. [Google Scholar] [CrossRef] [Green Version]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Ben Halim, N.; Ben Alaya Bouafif, N.; Romdhane, L.; Kefi Ben Atig, R.; Chouchane, I.; Bouyacoub, Y.; Arfa, I.; Cherif, W.; Nouira, S.; Talmoudi, F.; et al. Consanguinity, endogamy, and genetic disorders in Tunisia. J. Community Genet. 2013, 4, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Romdhane, L.; Mezzi, N.; Hamdi, Y.; El-Kamah, G.; Barakat, A.; Abdelhak, S. Consanguinity and Inbreeding in Health and Disease in North African Populations. Annu. Rev. Genom. Hum. Genet. 2019, 20, 155–179. [Google Scholar] [CrossRef]
- Lake, R.J.; Fan, H.Y. Structure, function and regulation of CSB: A multi-talented gymnast. Mech. Ageing Dev. 2013, 134, 202–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frontini, M.; Proietti-De-Santis, L. Interaction between the Cockayne syndrome B and p53 proteins: Implications for aging. Aging 2012, 4, 89–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okur, M.N.; Lee, J.H.; Osmani, W.; Kimura, R.; Demarest, T.G.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 2020, 48, 2473–2485. [Google Scholar] [CrossRef]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.M.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin-proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef] [PubMed]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEBJ. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2334–2346. [Google Scholar] [CrossRef] [Green Version]
- Epanchintsev, A.; Costanzo, F.; Rauschendorf, M.A.; Caputo, M.; Ye, T.; Donnio, L.M.; Proietti-de-Santis, L.; Coin, F.; Laugel, V.; Egly, J.M. Cockayne’s Syndrome A and B Proteins Regulate Transcription Arrest after Genotoxic Stress by Promoting ATF3 Degradation. Mol. Cell 2017, 68, 1054–1066.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epanchintsev, A.; Rauschendorf, M.-A.; Costanzo, F.; Calmels, N.; Obringer, C.; Sarasin, A.; Coin, F.; Laugel, V.; Egly, J.-M. Defective transcription of ATF3 responsive genes, a marker for Cockayne Syndrome. Sci. Rep. 2020, 10, 1105. [Google Scholar] [CrossRef] [Green Version]
- Crochemore, C.; Fernández-Molina, C.; Montagne, B.; Salles, A.; Ricchetti, M. CSB promoter downregulation via histone H3 hypoacetylation is an early determinant of replicative senescence. Nat. Commun. 2019, 10, 5576. [Google Scholar] [CrossRef] [Green Version]
- Cleaver, J.E.; Hefner, E.; Laposa, R.R.; Karentz, D.; Marti, T. Cockayne syndrome exhibits dysregulation of p21 and other gene products that may be independent of transcription-coupled repair. Neuroscience 2007, 145, 1300–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citterio, E.; Van Den Boom, V.; Schnitzler, G.; Kanaar, R.; Bonte, E.; Kingston, R.E.; Hoeijmakers, J.H.; Vermeulen, W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 2000, 20, 7643–7653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le May, N.; Mota-Fernandes, D.; Vélez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Limbo, O.; Fei, J.; Chen, L.; Kim, B.; Luo, J.; Chong, J.; Conaway, R.C.; Conaway, J.W.; Ranish, J.A.; et al. Regulation of the Rhp26ERCC6/CSB chromatin remodeler by a novel conserved leucine latch motif. Proc. Natl. Acad. Sci. USA 2014, 111, 18566–18571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, Y.; Tanaka, K.; Saijo, M. The C-terminal Region and SUMOylation of Cockayne Syndrome Group B Protein Play Critical Roles in Transcription-coupled Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Sonmez, F.M.; Celep, F.; Ugur, S.A. Severe Form of Cockayne Syndrome With Varying Clinical Presentation and No Photosensitivity in a Family. J. Child Neurol. 2006, 21, 333–337. [Google Scholar] [CrossRef]
- Colella, S.; Nardo, T.; Mallery, D.; Borrone, C.; Ricci, R.; Ruffa, G.; Lehmann, A.R.; Stefanini, M. Alterations in the CSB gene in three Italian patients with the severe form of Cockayne syndrome (CS) but without clinical photosensitivity. Hum. Mol. Genet. 1999, 8, 935–941. [Google Scholar] [CrossRef] [Green Version]
- Balogun, S.A.; Balogun, R.A.; Evans, J. Age-related differences in renal function at onset of renal replacement therapy in chronic kidney disease stage 5 patients. QJM Mon. J. Assoc. Physicians 2006, 99, 595–599. [Google Scholar] [CrossRef]
- Chatre, L.; Biard, D.S.F.; Sarasin, A.; Ricchetti, M. Reversal of mitochondrial defects with CSB-dependent serine protease inhibitors in patient cells of the progeroid Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, E2910–E2919. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | CS10 | CS12 | CD14 |
|---|---|---|---|
| Sex | M | F | M |
| Reference sequence | NM_000124.3 | NM_000124.3 | NM_000124.3 |
| Homozygous ERCC6 mutation | c.3156dup | c.3156dup | c.3156dup |
| Protein modification | p.(Arg1053Thr*8) | p.(Arg1053Thr*8) | p.(Arg1053Thr*8) |
| Geographic origin | Northwest | Northwest | Northwest |
| Consanguinity/Endogamy | Consanguineous | Consanguineous | Consanguineous |
| Range age at diagnosis (years) | 2–5 | 6–8 | 6–8 |
| Age at first symptoms(months) | Birth | Birth | 9 months |
| First symptoms | Growth delay | Growth delay | Psychomotor delay |
| Prenatal abnormalities | |||
| IUGR | NA | NA | NA |
| Microcephaly | + | + | + |
| Cerebellar hypoplasia | NA | NA | NA |
| Oligoamnios | NA | NA | NA |
| Birth findings | |||
| Birth weight (g) | 2550 | 2300 | 2950 |
| Birth height (cm) | 45 | 45 | 48 |
| Head circumference at birth (cm) | 33 | 32 | 32 |
| Postnatally findings (years) | 2–5 | 6–8 | −8 |
| Weight (kg) | 6 (−3 SD) | 8 (−4 SD) | 8 (−3 SD) |
| Height (cm) | 70 (−2 SD) | 82 (−6 SD) | 76 (−3 SD) |
| Head circumference (cm) | 41 (−2 SD) | 40 (−8.5 SD) | 42 (−3 SD) |
| Dysmorphism | |||
| Enophtalmia | + | + | + |
| Thin skin | − | + | + |
| Bird-like nose | + | + | + |
| Neurological findings | |||
| Microcephaly | + | + | + |
| Psychomotor delay | + | + | + |
| Independent sitting (months) | 8 | 5 | Not acquired |
| Independent walking (years) | 1.5 | 4 | Not acquired |
| Mental retardation | + | + | + |
| Limb spasticity | + | + | + |
| Retractions | − | + | + |
| Pyramidal signs | + | + | + |
| Neurogenic signs | − | + | + |
| Ataxia | + | + | − |
| Extrapyramidal signs | − | − | − |
| Epilepsy | − | − | − |
| Behavioral abnormalities | − | − | − |
| Ophthalmological findings | + | − | − |
| Cataract | − | − | + |
| Optic atrophy | − | + | − |
| Pigmentary retinopathy | |||
| Otolaryngological findings | |||
| Sensorineural deafness | + | + | + |
| Auditory evoked response | 60/70 dB | 60 dB (right ear)/no response for the left ear | NA |
| Dermatological findings | |||
| Photosensitivity | + | + | − |
| Eczema | − | − | − |
| Thin skin | − | + | − |
| Pigmentation abnormalities | − | − | + |
| Hair abnormalities | − | − | − |
| Nail abnormalities | − | − | − |
| Dental abnormalities | |||
| Caries | + | + | + |
| Tooth enamel abnormalities | − | + | + |
| Morphological teeth abnormalities | − | + | + |
| Laboratory findings | |||
| AST (NV < 40 U/L) | 65 | 108 | 45 |
| ALT (NV < 40 U/L) | 77 | 180 | 80 |
| Creatinine (NV50-110 µmol/L) | 22 | 20 | 31 |
| Imaging findings | |||
| Calcifications | + | + | + |
| Hypomyelination | + | − | + |
| Cerebellar atrophy | + | + | + |
| Brainstem atrophy | + | + | + |
| Nerve conduction velocities | NA | (SPE. D) 23.5 m/s slowed | (SPE. D) 16 m/s slowed |
| Neurophysiological findings (ENMG Test) | NA | Sensory and motor demyelinating polyneuropathy | Sensory and motor demyelinating polyneuropathy |
| Others Findings | Cryptorchidia, toxoplasmosis during pregnancy | − | Cryptorchidia, kyphosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zayoud, K.; Kraoua, I.; Chikhaoui, A.; Calmels, N.; Bouchoucha, S.; Obringer, C.; Crochemore, C.; Najjar, D.; Zarrouk, S.; Miladi, N.; et al. Identification and Characterization of a Novel Recurrent ERCC6 Variant in Patients with a Severe Form of Cockayne Syndrome B. Genes 2021, 12, 1922. https://doi.org/10.3390/genes12121922
Zayoud K, Kraoua I, Chikhaoui A, Calmels N, Bouchoucha S, Obringer C, Crochemore C, Najjar D, Zarrouk S, Miladi N, et al. Identification and Characterization of a Novel Recurrent ERCC6 Variant in Patients with a Severe Form of Cockayne Syndrome B. Genes. 2021; 12(12):1922. https://doi.org/10.3390/genes12121922
Chicago/Turabian StyleZayoud, Khouloud, Ichraf Kraoua, Asma Chikhaoui, Nadège Calmels, Sami Bouchoucha, Cathy Obringer, Clément Crochemore, Dorra Najjar, Sinda Zarrouk, Najoua Miladi, and et al. 2021. "Identification and Characterization of a Novel Recurrent ERCC6 Variant in Patients with a Severe Form of Cockayne Syndrome B" Genes 12, no. 12: 1922. https://doi.org/10.3390/genes12121922