
The Effects of Sequence Length and Composition of Random Sequence Peptides on the Growth of E. coli Cells


Abstract
:1. Introduction
2. Materials and Methods
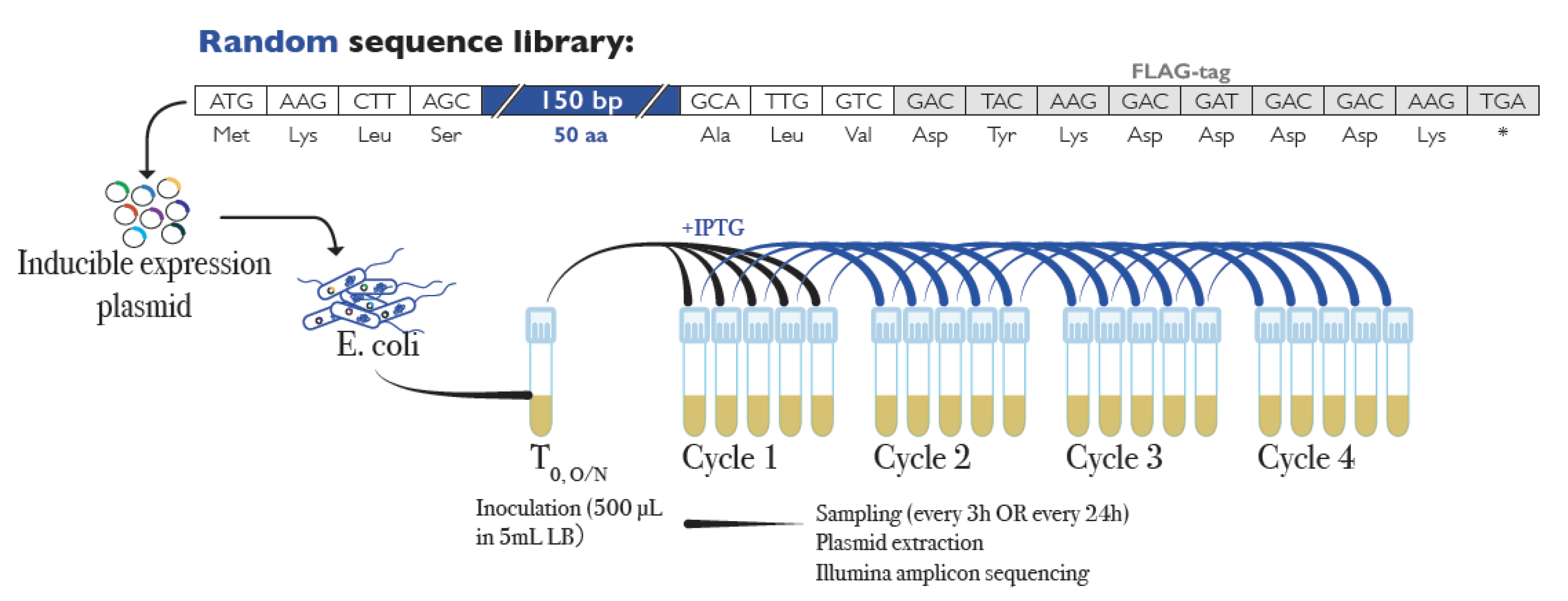
2.1. Library and Replication Experiment
2.2. Analysis Pipeline
2.3. Database Generation
2.4. Sequence Features
2.5. Mapping of Reads to Full Database
2.6. Frequency Change Determination and Group Assignment
3. Results
3.1. Replication with Diluted Library
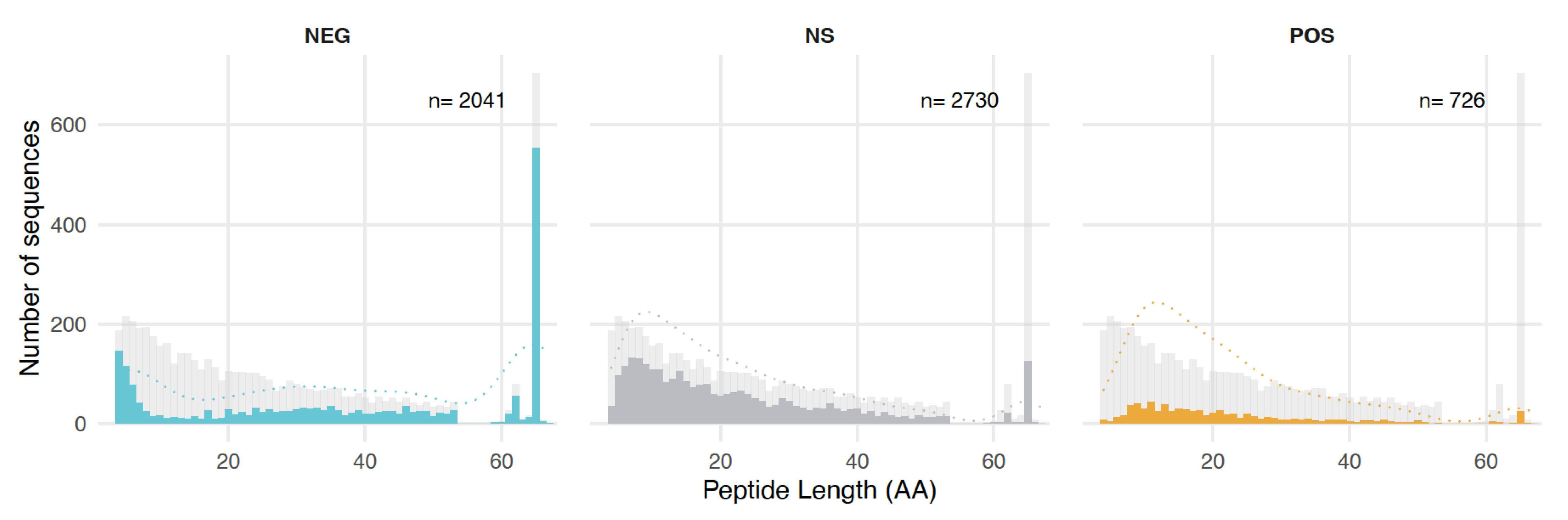
3.2. Characterization of the Sequences in the Random Clone Library
3.3. Frequency Changes in Clones during the Growth Experiments
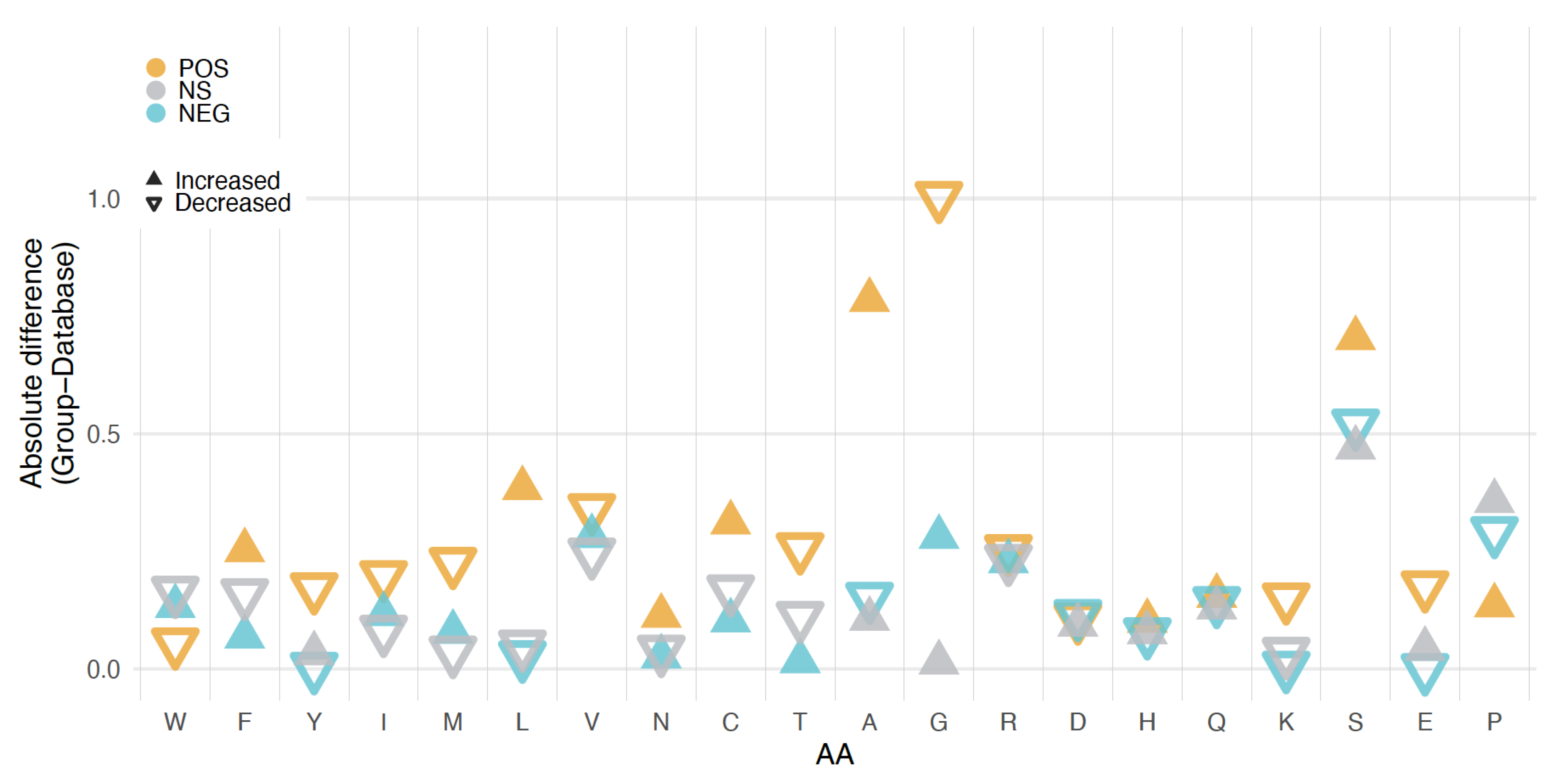
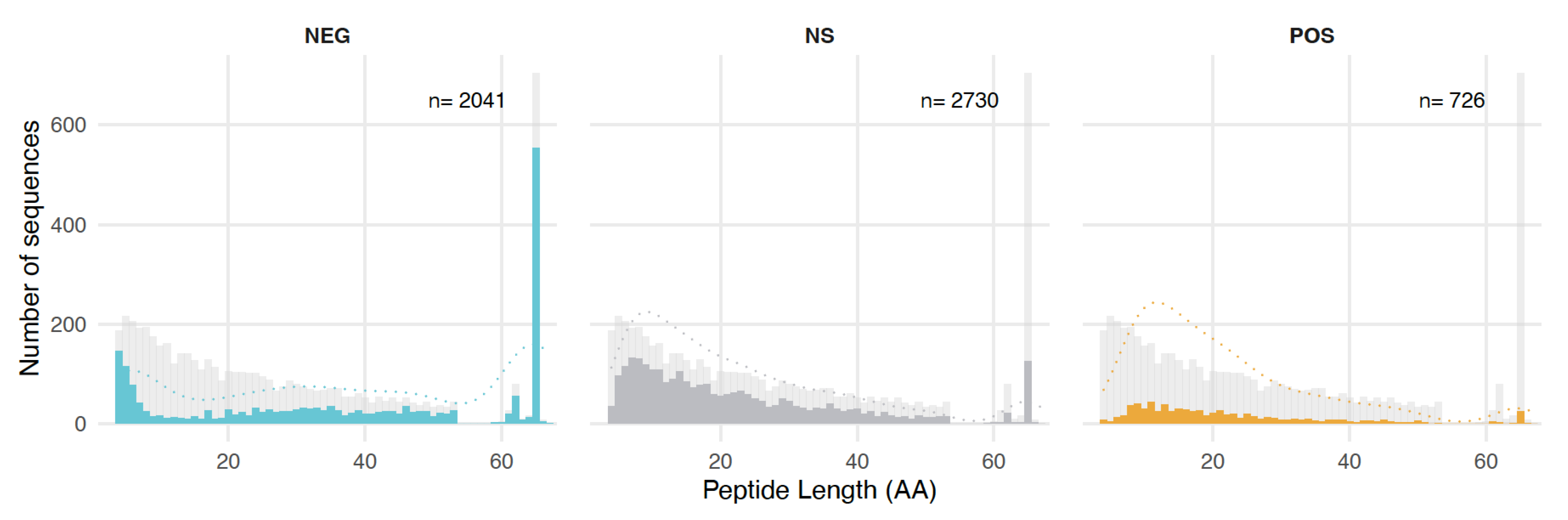
3.4. Length, GC Content, and Amino Acid Composition Dependence
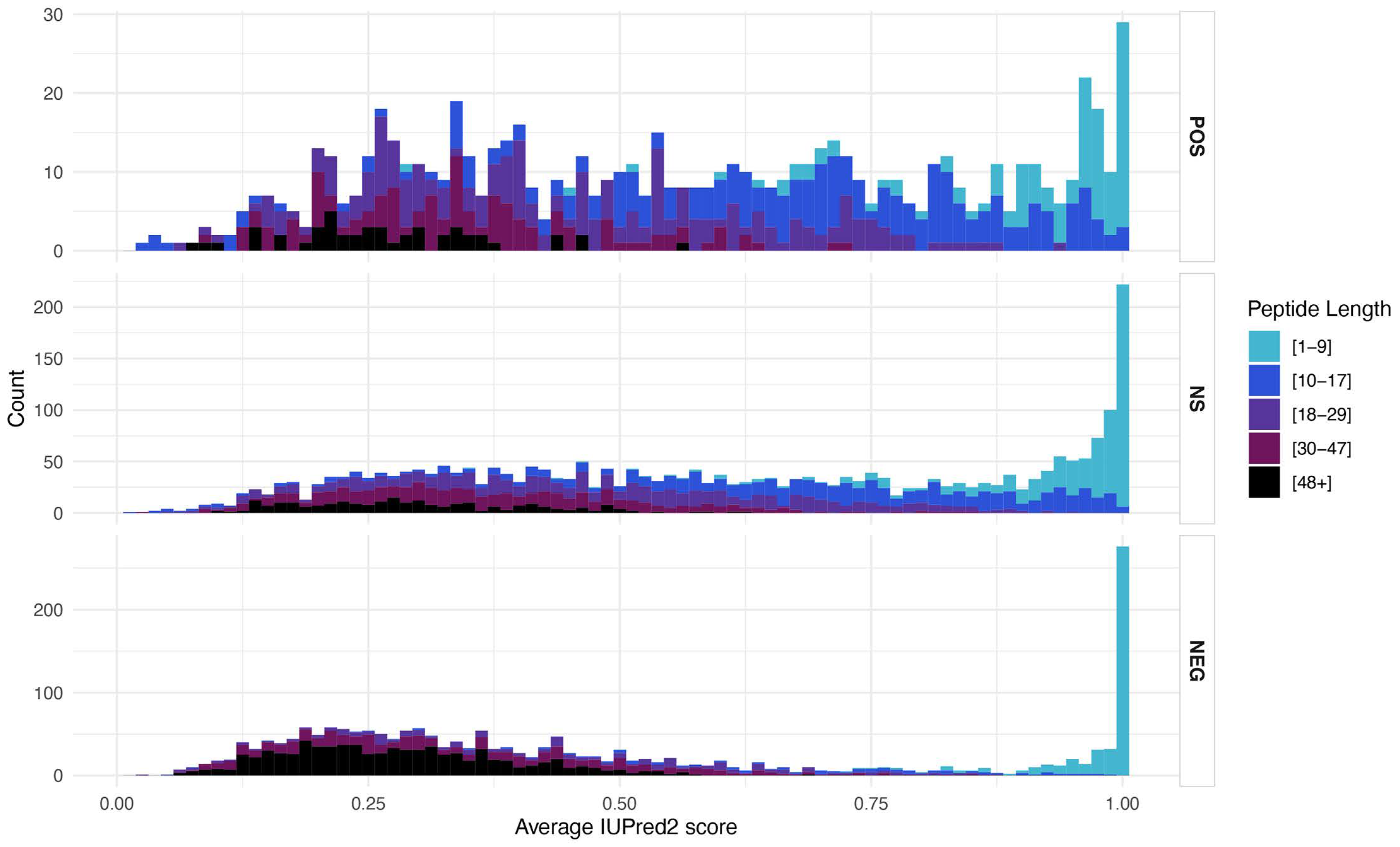
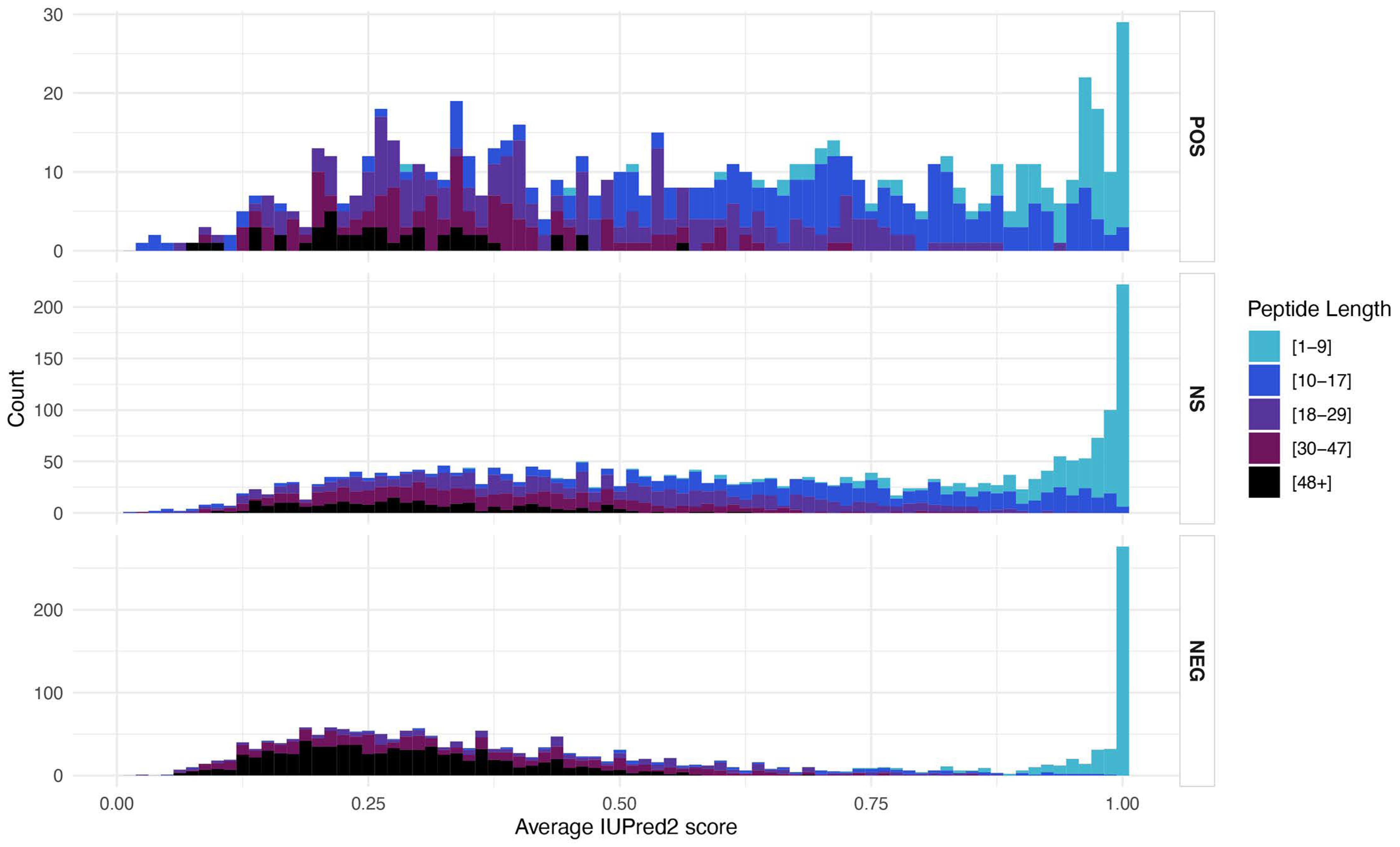
3.5. Structural Features
4. Discussion
4.1. New Analysis Pipeline
4.2. Clone Effects
4.3. Negative Effects of Vector Coded Amino Acids
4.4. Protein Structure Correlations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tautz, D.; Domazet-Loso, T. The evolutionary origin of orphan genes. Nat. Rev. Genet. 2011, 12, 692–702. [Google Scholar] [CrossRef]
- Chen, S.; Krinsky, B.H.; Long, M. New genes as drivers of phenotypic evolution. Nat. Rev. Genet. 2013, 14, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlötterer, C. Genes from scratch—The evolutionary fate of de novo genes. Trends Genet. 2015, 31, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLysaght, A.; Guerzoni, D. New genes from non-coding sequence: The role of de novo protein-coding genes in eukaryotic evolutionary innovation. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Oss, S.B.; Carvunis, A.-R. De novo gene birth. PLoS Genet. 2019, 15, e1008160. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.I.; Jerlström-Hultqvist, J.; Näsvall, J. Evolution of New Functions De Novo and from Preexisting Genes. Cold Spring Harb. Perspect. Biol. 2015, 7, a017996. [Google Scholar] [CrossRef]
- Neme, R.; Tautz, D. Phylogenetic patterns of emergence of new genes support a model of frequent de novo evolution. BMC Genom. 2013, 14, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neme, R.; Tautz, D. Fast turnover of genome transcription across evolutionary time exposes entire non-coding DNA to de novo gene emergence. eLife 2016, 5, e09977. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Orera, J.; Messeguer, X.; Subirana, J.; Alba, M.M. Long non-coding RNAs as a source of new peptides. eLife 2014, 3, e03523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, B.A.; Foy, S.G.; Neme, R.; Masel, J. Young genes are highly disordered as predicted by the preadaptation hypothesis of de novo gene birth. Nat. Ecol. Evol. 2017, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pavesi, A.; Magiorkinis, G.; Karlin, D.G. Viral Proteins Originated De Novo by Overprinting Can Be Identified by Codon Usage: Application to the “Gene Nursery” of Deltaretroviruses. PLoS Comput. Biol. 2013, 9, e1003162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, J.E.; Willis, S.M.; Nelson, P.G.; Weibel, C.; Kosinski, L.J.; Masel, J. Universal and taxon-specific trends in protein sequences as a function of age. eLife 2021, 10, e57347. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Yang, T.; Li, G.; Chen, J.; Gschwend, A.R.; Yu, Y.; Hou, G.; Zi, J.; Zhou, R. Rapid evolution of protein diversity by de novo origination in Oryza. Nat. Ecol. Evol. 2019, 3, 679–690. [Google Scholar] [CrossRef]
- Weisman, C.M.; Murray, A.W.; Eddy, S.R. Many, but not all, lineage-specific genes can be explained by homology detection failure. PLoS Biol. 2020, 18, e3000862. [Google Scholar] [CrossRef]
- Heinen, T.J.; Staubach, F.; Häming, D.; Tautz, D. Emergence of a New Gene from an Intergenic Region. Curr. Biol. 2009, 19, 1527–1531. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Bekpen, C.; Künzel, S.; Keshavarz, M.; Krebs-Wheaton, R.; Skrabar, N.; Ullrich, K.K.; Tautz, D. A de novo evolved gene in the house mouse regulates female pregnancy cycles. eLife 2019, 8, e44392. [Google Scholar] [CrossRef]
- Cai, J.; Zhao, R.; Jiang, H.; Wang, W. De Novo Origination of a New Protein-Coding Gene in Saccharomyces cerevisiae. Genetics 2008, 179, 487–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Dong, Y.; Jiang, Y.; Jiang, H.; Cai, J.; Wang, W. A de novo originated gene depresses budding yeast mating pathway and is repressed by the protein encoded by its antisense strand. Cell Res. 2010, 20, 408–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reinhardt, J.; Wanjiru, B.M.; Brant, A.T.; Saelao, P.; Begun, D.J.; Jones, C.D. De Novo ORFs in Drosophila Are Important to Organismal Fitness and Evolved Rapidly from Previously Non-coding Sequences. PLoS Genet. 2013, 9, e1003860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepanov, V.G.; Fox, G.E. Stress-Driven In Vivo Selection of a Functional Mini-Gene from a Randomized DNA Library Expressing Combinatorial Peptides in Escherichia coli. Mol. Biol. Evol. 2007, 24, 1480–1491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopp, M.; Gudmundsdottir, J.S.; Nilsson, T.; König, F.; Warsi, O.; Rajer, F.; Ädelroth, P.; Andersson, D.I. De Novo Emergence of Peptides That Confer Antibiotic Resistance. mBio 2019, 10, e00837-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knopp, M.; Babina, A.M.; Gudmundsdóttir, J.S.; Douglass, M.V.; Trent, M.S.; Andersson, D.I. A novel type of colistin resistance genes selected from random sequence space. PLoS Genet. 2021, 17, e1009227. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Clancy, M.A.; Carvalho, R.F.; Elliott, K.; Folta, K.M. Identification of Novel Growth Regulators in Plant Populations Expressing Random Peptides. Plant Physiol. 2017, 175, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, A.D.; Szostak, J.W. Functional proteins from a random-sequence library. Nat. Cell Biol. 2001, 410, 715–718. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Saelao, P.; Jones, C.D.; Begun, D.J. Origin and Spread of de Novo Genes in Drosophila melanogaster Populations. Science 2014, 343, 769–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmieri, N.; Kosiol, C.; Schlötterer, C. The life cycle of Drosophila orphan genes. eLife 2014, 3, e01311. [Google Scholar] [CrossRef] [Green Version]
- Neme, R.; Tautz, D. Evolution: Dynamics of De Novo Gene Emergence. Curr. Biol. 2014, 24, R238–R240. [Google Scholar] [CrossRef] [Green Version]
- Durand, É.; Gagnon-Arsenault, I.; Hallin, J.; Hatin, I.; Dubé, A.K.; Nielly-Thibault, L.; Namy, O.; Landry, C.R. Turnover of ribosome-associated transcripts from de novo ORFs produces gene-like characteristics available for de novo gene emergence in wild yeast populations. Genome Res. 2019, 29, 932–943. [Google Scholar] [CrossRef]
- Neme, R.; Amador, C.; Yildirim, B.; McConnell, E.; Tautz, D. Random sequences are an abundant source of bioactive RNAs or peptides. Nat. Ecol. Evol. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Weisman, C.; Eddy, S.R. Gene Evolution: Getting Something from Nothing. Curr. Biol. 2017, 27, R661–R663. [Google Scholar] [CrossRef] [Green Version]
- Knopp, M.; Andersson, D.I. No beneficial fitness effects of random peptides. Nat. Ecol. Evol. 2018, 2, 1046–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tautz, D.; Neme, R. Reply to ‘No beneficial fitness effects of random peptides’. Nat. Ecol. Evol. 2018, 2, 1048. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Walsh, I.; Seno, F.; Tosatto, S.C.; Trovato, A. PASTA 2.0: An improved server for protein aggregation prediction. Nucleic Acids Res. 2014, 42, W301–W307. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Salit, M.L.; Levy, S.F. Unbiased Fitness Estimation of Pooled Barcode or Amplicon Sequencing Studies. Cell Syst. 2018, 7, 521–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heames, B.; Schmitz, J.; Bornberg-Bauer, E. A Continuum of Evolving De Novo Genes Drives Protein-Coding Novelty in Drosophila. J. Mol. Evol. 2020, 88, 382–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basile, W.; Sachenkova, O.; Light, S.; Elofsson, A. High GC content causes orphan proteins to be intrinsically disordered. PLoS Comput. Biol. 2017, 13, e1005375. [Google Scholar] [CrossRef]
- Yuedong, Y.; Cao, Z.; Yang, Y.; Wang, C.-L.; Su, Z.-D.; Zhao, Y.-W.; Wang, J.-H.; Zhou, Y. Natural protein sequences are more intrinsically disordered than random sequences. Cell. Mol. Life Sci. 2016, 73, 2949–2957. [Google Scholar] [CrossRef] [Green Version]
- Campen, A.; Williams, R.; Brown, C.; Meng, J.; Uversky, V.; Dunker, A. TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder. Protein Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvunis, A.-R.; Rolland, T.; Wapinski, I.; Calderwood, M.; Yildirim, M.; Simonis, N.; Charloteaux, B.; Hidalgo, C.; Barbette, J.; Santhanam, B.; et al. Proto-genes and de novo gene birth. Nat. Cell Biol. 2012, 487, 370–374. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.F.; Ullrich, K.K.; Bornberg-Bauer, E. Incipient de novo genes can evolve from frozen accidents that escaped rapid transcript turnover. Nat. Ecol. Evol. 2018, 2, 1626–1632. [Google Scholar] [CrossRef]
- Mittal, P.; Brindle, J.; Stephen, J.; Plotkin, J.B.; Kudla, G. Codon usage influences fitness through RNA toxicity. Proc. Natl. Acad. Sci. USA 2018, 115, 8639–8644. [Google Scholar] [CrossRef] [Green Version]
- Walsh, I.M.; Bowman, M.A.; Santarriaga, I.F.S.; Rodriguez, A.; Clark, P.L. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl. Acad. Sci. USA 2020, 117, 3528–3534. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Rinas, U. Recombinant protein production associated growth inhibition results mainly from transcription and not from translation. Microb. Cell Factories 2020, 19, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tretyachenko, V.; Vymětal, J.; Bednárová, L.; Kopecký, V.; Hofbauerova, K.; Jindrová, H.; Hubálek, M.; Souček, R.; Konvalinka, J.; Vondrášek, J.; et al. Random protein sequences can form defined secondary structures and are well-tolerated in vivo. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Creixell, P.; Schoof, E.M.; Tan, C.S.H.; Linding, R. Mutational properties of amino acid residues: Implications for evolvability of phosphorylatable residues. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 2584–2593. [Google Scholar] [CrossRef] [Green Version]
- Bhave, D.; Tautz, D. Effects of the expression of random sequence clones on growth and transcriptome regulation in Escherichia coli. bioRxiv 2021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exp 1 | Cycle Length/Experiment Length (Replicates) | N 2 | POS 3 | NEG 3 | NS 3 | Range Log2-Fold Change (Average) | Empty Vector Log2-Fold Change 4 |
|---|---|---|---|---|---|---|---|
| 1 | 3 h/1 day (n = 10) | 5625 | 0.11 | 0.36 | 0.53 | −8.0 to 2.7 (−1.1) | 1.1 |
| 2 | 3 h/1 day (n = 10) | 5606 | 0.17 | 0.43 | 0.41 | −7.7 to 2.2 (−1.2) | 0.5 |
| 3 | 3 h/1 day (n = 10) | 5638 | 0.18 | 0.40 | 0.42 | −7.8 to 2.7 (−1.1) | 0.4 |
| 4 | 24 h/4 days (n = 8) | 5623 | 0.14 | 0.30 | 0.56 | −5.2 to 5.2 (−0.5) | 0.1 |
| 5 | 24 h/4 days (n = 10) | 5596 | 0.10 | 0.26 | 0.64 | −5.4 to 5.0 (−0.6) | −1.7 |
| 6 | 24 h/4 days (n = 10) | 5632 | 0.26 | 0.41 | 0.32 | −5.9 to 2.2 (−0.7) | −1.1 |
| 7 | 24 h/4 days (n = 5) | 5623 | 0.07 | 0.28 | 0.65 | −7.2 to 4.0 (−0.9) | −0.2 |
| 8 | 24 h/4 days (n = 5) | 5689 | 0.27 | 0.46 | 0.27 | −11.2 to 1.4 (−1.6) | −0.4 |
| 9 | 3 h/1 day (n = 5)/diluted library | 5651 | 0.16 | 0.32 | 0.51 | −8.4 to 5.6 (−0.7) | −0.7 |
| All experiments averages 5: | |||||||
| All clones | 5621 | 0.16 | 0.36 | 0.48 | |||
| Clones with 4aa ORF | 200 | 0.04 | 0.73 | 0.18 | |||
| Clones with 5aa ORF | 221 | 0.02 | 0.52 | 0.44 | |||
| Clones with 6aa ORF | 209 | 0.06 | 0.37 | 0.56 | |||
| Clones with FLAG sequence | 638 | 0.03 | 0.77 | 0.17 | |||
| Clones with FLAG + 1 sequence | 129 | 0.03 | 0.68 | 0.20 | |||
| Clones with FLAG + 2 sequence | 126 | 0.05 | 0.64 | 0.23 | |||
| Clones 48+ aa without FLAG | 237 | 0.06 | 0.55 | 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castro, J.F.; Tautz, D. The Effects of Sequence Length and Composition of Random Sequence Peptides on the Growth of E. coli Cells. Genes 2021, 12, 1913. https://doi.org/10.3390/genes12121913
Castro JF, Tautz D. The Effects of Sequence Length and Composition of Random Sequence Peptides on the Growth of E. coli Cells. Genes. 2021; 12(12):1913. https://doi.org/10.3390/genes12121913
Chicago/Turabian StyleCastro, Johana F., and Diethard Tautz. 2021. "The Effects of Sequence Length and Composition of Random Sequence Peptides on the Growth of E. coli Cells" Genes 12, no. 12: 1913. https://doi.org/10.3390/genes12121913
APA StyleCastro, J. F., & Tautz, D. (2021). The Effects of Sequence Length and Composition of Random Sequence Peptides on the Growth of E. coli Cells. Genes, 12(12), 1913. https://doi.org/10.3390/genes12121913