
Association of a Total Cholesterol Polygenic Score with Cholesterol Levels and Pathological Biomarkers across the Alzheimer’s Disease Spectrum

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. The Meta-Analysis Summary Data
2.2. Study Populations
2.2.1. PREVENT-AD
2.2.2. ADNI
2.2.3. ROSMAP
2.3. Genetic Data
2.3.1. Quality Control
2.3.2. Imputation
2.3.3. Merging ADNI Data
2.4. TC-PGS
2.5. TC and Hypercholesterolemia Measurements
2.6. CSF Measurements
2.7. PET Imaging
2.8. Amyloid Positivity Status
2.9. Statistical Analyses
2.9.1. Descriptive
2.9.2. TC Levels and p-Value Thresholding
2.9.3. Hypercholesterolemia ROC Analyses
2.9.4. CSF and PET Linear Regression Analyses
2.9.5. Risk of AD and Cognitive Impairment Logistic Regression Analyses
2.9.6. Conversion Rate
3. Results
3.1. Cohort Characteristics
Global Lipids Genetics Consortium
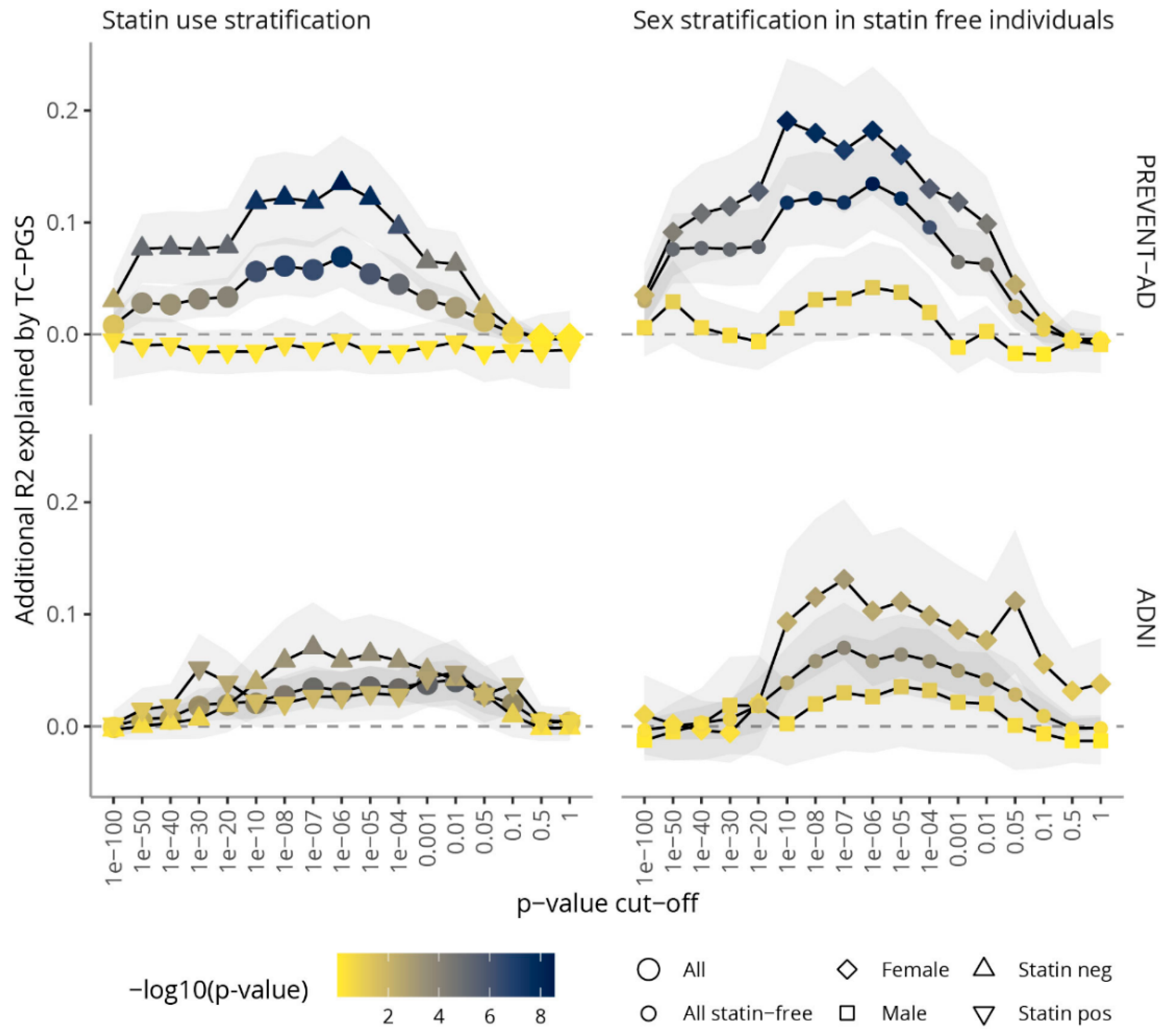
3.2. Amount of Variance Explained in TC Blood Levels by TC-PGS
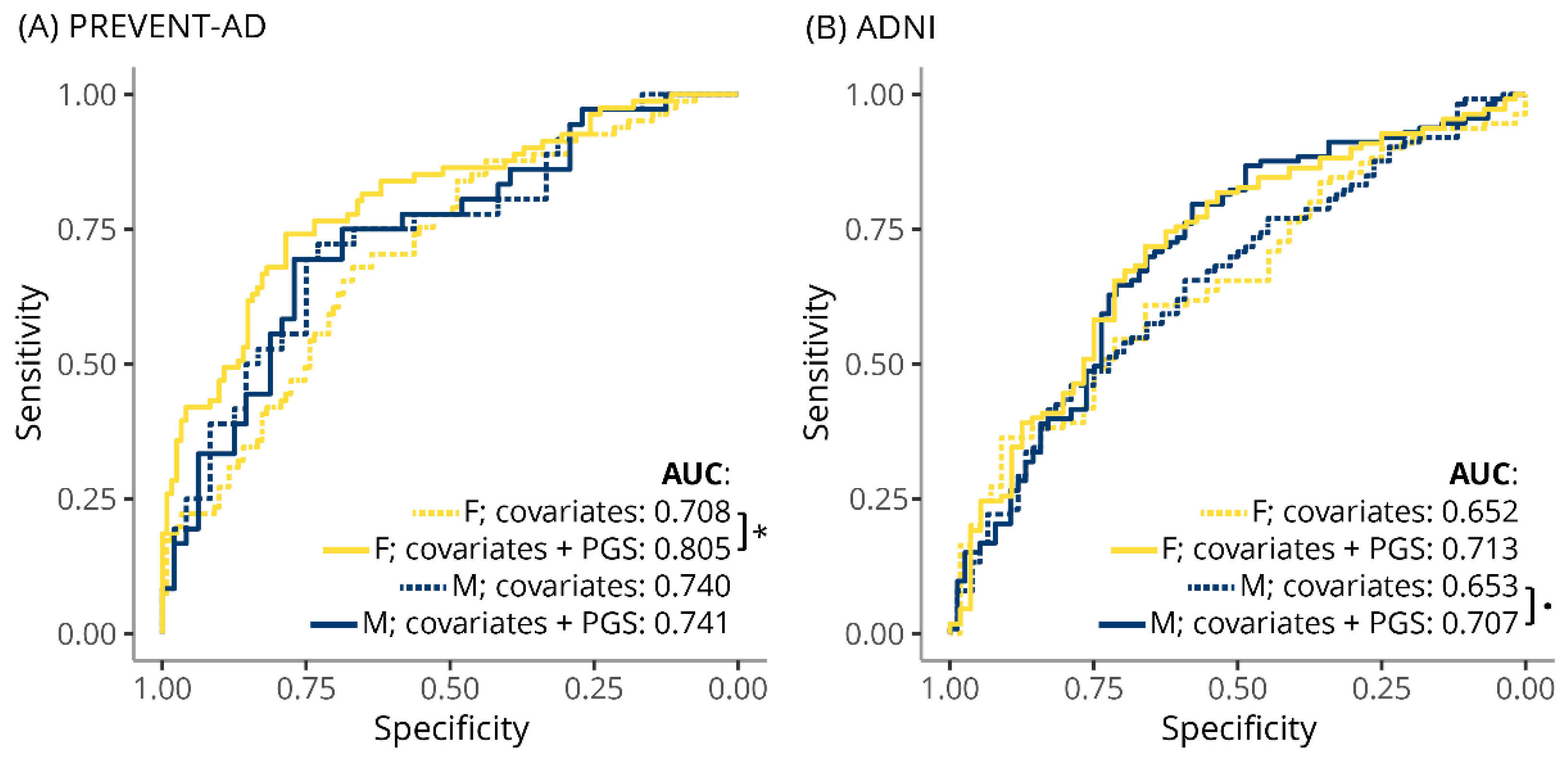
3.3. TC-PGS Predicts Hypercholesterolemia
3.4. TC-PGS Does Not Associate with Amyloid Pathology
3.5. TC-PGS Does Not Associate with TAU Pathology
3.6. TC-PGS Does Not Associate with Markers of Neurodegeneration
3.7. TC-PGS Does Not Associate with Increased Risk of Becoming Aβ(+)
3.8. TC-PGS Does Not Associate with Cognition in Aβ(+) Individuals
4. Discussion
4.1. TC-PGS and TC Levels
4.2. TC-PGS and AD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. 2017 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 13, 325–373. [Google Scholar] [CrossRef]
- Ridge, P.G.; Hoyt, K.B.; Boehme, K.; Mukherjee, S.; Crane, P.K.; Haines, J.L.; Mayeux, R.; Farrer, L.; Pericak-Vance, M.A.; Schellenberg, G.D.; et al. Assessment of the genetic variance of late-onset Alzheimer’s disease. Neurobiol. Aging 2016, 41, 200.e13–200.e20. [Google Scholar] [CrossRef] [PubMed]
- Reiman, E.M.; Chen, K.; Langbaum, J.B.; Lee, W.; Reschke, C.; Bandy, D.; Alexander, G.E.; Caselli, R.J. Higher serum total cholesterol levels in late middle age are associated with glucose hypometabolism in brain regions affected by Alzheimer’s disease and normal aging. NeuroImage 2010, 49, 169–176. [Google Scholar] [CrossRef]
- Solomon, A.; Kivipelto, M.; Wolozin, B.; Zhou, J.; Whitmer, R.A. Midlife Serum Cholesterol and Increased Risk of Alzheimer’s and Vascular Dementia Three Decades Later. Dement. Geriatr. Cogn. Disord. 2009, 28, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Toro, P.; Degen, C.; Pierer, M.; Gustafson, D.; Schröder, J.; Schönknecht, P. Cholesterol in mild cognitive impairment and Alzheimer’s disease in a birth cohort over 14 years. Eur. Arch. Psychiatry Clin. Neurosci. 2014, 264, 485–492. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissinen, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. BMJ 2001, 322, 1447–1451. [Google Scholar] [CrossRef]
- Kivipelto, M.; Helkala, E.-L.; Laakso, M.; Hänninen, T.; Hallikainen, M.; Alhainen, K.; Iivonen, S.; Mannermaa, A.; Tuomilehto, J.; Nissinen, A.; et al. Apolipoprotein E ϵ4 Allele, Elevated Midlife Total Cholesterol Level, and High Midlife Systolic Blood Pressure Are Independent Risk Factors for Late-Life Alzheimer Disease. Ann. Intern. Med. 2002, 137, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Ngandu, T.; Fratiglioni, L.; Viitanen, M.; Kåreholt, I.; Winblad, B.; Helkala, E.-L.; Tuomilehto, J.; Soininen, H.; Nissinen, A. Obesity and Vascular Risk Factors at Midlife and the Risk of Dementia and Alzheimer Disease. Arch. Neurol. 2005, 62, 1556–1560. [Google Scholar] [CrossRef]
- Li, G.; Shofer, J.B.; Kukull, W.A.; Peskind, E.R.; Tsuang, D.W.; Breitner, J.; McCormick, W.; Bowen, J.D.; Teri, L.; Schellenberg, G.D.; et al. Serum cholesterol and risk of Alzheimer disease: A community-based cohort study. Neurology 2005, 65, 1045–1050. [Google Scholar] [CrossRef]
- Tan, Z.S.; Seshadri, S.; Beiser, A.; Wilson, P.W.F.; Kiel, D.; Tocco, M.; D’Agostino, R.B.; Wolf, P.A. Plasma Total Cholesterol Level as a Risk Factor for Alzheimer Disease. Arch. Intern. Med. 2003, 163, 1053–1057. [Google Scholar] [CrossRef]
- Mielke, M.M.; Zandi, P.P.; Shao, H.; Waern, M.M.; Östling, S.; Guo, X.M.; Björkelund, C.; Lissner, L.; Skoog, I.M. The 32-year relationship between cholesterol and dementia from midlife to late life. Neurology 2010, 75, 1888–1895. [Google Scholar] [CrossRef]
- Pappolla, M.A.; Bryant-Thomas, T.; Herbert, D.; Pacheco, J.; Garcia, M.F.; Manjon, M.; Gironès, X.; Henry, T.; Matsubara, E.; Zambon, D.; et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 2003, 61, 199–205. [Google Scholar] [CrossRef]
- Köbe, T.; Gonneaud, J.; Binette, A.P.; Meyer, P.-F.; McSweeney, M.; Rosa-Neto, P.; Breitner, J.C.S.; Poirier, J.; Villeneuve, S. For the Presymptomatic Evaluation of Experimental or Novel Treatments for Alzheimer Disease (PREVENT-AD) Research Group Association of Vascular Risk Factors With β-Amyloid Peptide and Tau Burdens in Cognitively Unimpaired Individuals and Its Interaction With Vascular Medication Use. JAMA Netw. Open 2020, 3, e1920780. [Google Scholar] [CrossRef]
- Reed, B.; Villeneuve, S.; Mack, W.; DeCarli, C.; Chui, H.C.; Jagust, W. Associations Between Serum Cholesterol Levels and Cerebral Amyloidosis. JAMA Neurol. 2014, 71, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Lesser, G.; Kandiah, K.; Libow, L.; Likourezos, A.; Breuer, B.; Marin, D.; Mohs, R.; Haroutunian, V.; Neufeld, R. Elevated Serum Total and LDL Cholesterol in Very Old Patients with Alzheimer’s Disease. Dement. Geriatr. Cogn. Disord. 2001, 12, 138–145. [Google Scholar] [CrossRef]
- Lesser, G.T.; Haroutunian, V.; Purohit, D.P.; Beeri, M.S.; Schmeidler, J.; Honkanen, L.; Neufeld, R.; Libow, L.S. Serum Lipids Are Related to Alzheimer’s Pathology in Nursing Home Residents. Dement. Geriatr. Cogn. Disord. 2009, 27, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, M.; Zahiri, H.R.; Ceimo, J.; Cooper, K.; Gaul, W.; Connor, D.; Sparks, D.L. Is there a characteristic lipid profile in Alzheimer’s disease? J. Alzheimer’s Dis. 2005, 6, 585–589. [Google Scholar] [CrossRef]
- Mielke, M.M.; Zandi, P.P.; Sjogren, M.; Gustafson, D.; Ostling, S.; Steen, B.; Skoog, I. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology 2005, 64, 1689–1695. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Carrillo, M.C.; Forman, M.; DeSanti, S.; Miller, D.S.; Kozauer, N.; Petersen, R.C.; Randolph, C.; Knopman, D.S.; Smith, E.E.; et al. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2017, 3, 83–91. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Clifford, R.J., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Feldman, H.H.; Frisoni, G.B.; Hampel, H.; Jagust, W.J.; Johnson, K.A.; Knopman, D.S.; et al. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 2016, 87, 539–547. [Google Scholar] [CrossRef]
- The ENGAGE Consortium Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat. Genet. 2009, 41, 47–55. [CrossRef] [PubMed]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef]
- Teslovich, T.M.; Musunuru, K.; Smith, A.V.; Edmondson, A.C.; Stylianou, I.M.; Koseki, M.; Pirruccello, J.P.; Ripatti, S.; Chasman, D.I.; Willer, C.J.; et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010, 466, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Gatz, M.; Reynolds, C.A.; Fratiglioni, L.; Johansson, B.; Mortimer, J.A.; Berg, S.; Fiske, A.; Pedersen, N.L. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168–174. [Google Scholar] [CrossRef]
- Heller, D.A.; De Faire, U.; Pedersen, N.L.; Dahlén, G.; McClearn, G.E. Genetic and Environmental Influences on Serum Lipid Levels in Twins. N. Engl. J. Med. 1993, 328, 1150–1156. [Google Scholar] [CrossRef]
- Proitsi, P.; Lupton, M.; Velayudhan, L.; Newhouse, S.; Fogh, I.; Tsolaki, M.; Daniilidou, M.; Pritchard, M.R.; Kloszewska, I.; Soininen, H.; et al. Genetic Predisposition to Increased Blood Cholesterol and Triglyceride Lipid Levels and Risk of Alzheimer Disease: A Mendelian Randomization Analysis. PLoS Med. 2014, 11, e1001713. [Google Scholar] [CrossRef] [PubMed]
- Breitner, J.C.S.; Poirier, J.; Etienne, P.E.; Leoutsakos, J.M. Rationale and Structure for a New Center for Studies on Prevention of Alzheimer’s Disease (StoP-AD). J. Prev. Alzheimers Dis. 2016, 3, 236–242. [Google Scholar] [CrossRef]
- Bennett, D.A.; Buchman, A.S.; Boyle, P.A.; Barnes, L.L.; Wilson, R.S.; Schneider, J.A. Religious Orders Study and Rush Memory and Aging Project. J. Alzheimer’s Dis. 2018, 64, S161–S189. [Google Scholar] [CrossRef]
- Weale, M.E. Quality Control for Genome-Wide Association Studies. In MAP Kinase Signaling Protocols; Springer: Singapore, 2010; Volume 628, pp. 341–372. [Google Scholar]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.D.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Sudmant, P.H.; Rausch, T.; Gardner, E.J.; Handsaker, E.R.; Abyzov, A.; Huddleston, J.; Zhang, Y.; Ye, K.; Jun, G.; Fritz, M.H.-Y.; et al. An integrated map of structural variation in 2504 human genomes. Nature 2015, 526, 75–81. [Google Scholar] [CrossRef] [PubMed]
- The 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Delaneau, O.; Marchini, J.; Zagury, J.-F. A linear complexity phasing method for thousands of genomes. Nat. Methods 2011, 9, 179–181. [Google Scholar] [CrossRef]
- Durbin, R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014, 30, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.S.; de Andrade, M.; Tromp, G.; Kuivaniemi, H.; Epugh, E.; Namjou-Khales, B.; Mukherjee, S.; Jarvik, G.P.; Kottyan, L.; Eburt, A.; et al. Imputation and quality control steps for combining multiple genome-wide datasets. Front. Genet. 2014, 5, 370. [Google Scholar] [CrossRef]
- Meyer, P.-F.; Tremblay-Mercier, J.; Leoutsakos, J.; Madjar, C.; Lafaille-Maignan, M.-É.; Savard, M.; Rosa-Neto, P.; Poirier, J.; Etienne, P.; Breitner, J.; et al. INTREPAD. Neurology 2019, 92, e2070–e2080. [Google Scholar] [CrossRef] [PubMed]
- Bittner, T.; Zetterberg, H.; Teunissen, C.E.; Ostlund, R.E.; Militello, M.; Andreasson, U.; Hubeek, I.; Gibson, D.; Chu, D.C.; Eichenlaub, U.; et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1–42) in human cerebrospinal fluid. Alzheimer’s Dement. 2016, 12, 517–526. [Google Scholar] [CrossRef]
- Shaw, L.M.; Hansson, O.; Manuilova, E.; Masters, C.L.; Doecke, J.D.; Li, Q.-X.; Rutz, S.; Widmann, M.; Leinenbach, A.; Blennow, K. Method comparison study of the Elecsys® β-Amyloid (1–42) CSF assay versus comparator assays and LC-MS/MS. Clin. Biochem. 2019, 72, 7–14. [Google Scholar] [CrossRef] [PubMed]
- McSweeney, M.; Binette, A.P.; Meyer, P.-F.; Gonneaud, J.; Bedetti, C.; Ozlen, H.; Labonté, A.; Rosa-Neto, P.; Breitner, J.; Poirier, J.; et al. Intermediate flortaucipir uptake is associated with Aβ-PET and CSF tau in asymptomatic adults. Neurology 2020, 94, e1190–e1200. [Google Scholar] [CrossRef] [PubMed]
- Hansson, O.; Seibyl, J.; Stomrud, E.; Zetterberg, H.; Trojanowski, J.Q.; Bittner, T.; Lifke, V.; Corradini, V.; Eichenlaub, U.; Batrla, R.; et al. CSF biomarkers of Alzheimer’s disease concord with amyloid-β PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer’s Dement. 2018, 14, 1470–1481. [Google Scholar] [CrossRef]
- P Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: http://www.R-project.org (accessed on 18 January 2019).
- Dowle, M.; Srinivasan, A.; Gorecki, J.; Chirico, M.; Stetsenko, P.; Short, T.; Lianoglou, S.; Antonyan, E.; Bonsch, M.; Parsonage, H.; et al. Data.Table: Extension of ‘Data.Frame’. Available online: https://CRAN.R-project.org/package=data.table (accessed on 29 May 2020).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2”. 2019. Available online: https://CRAN.R-project.org/package=cowplot (accessed on 17 June 2020).
- Revelle, W. Psych: Procedures for Personality and Psychological Research; Northwestern University: Evanston, IL, USA, 2017; Available online: https://CRAN.R-project.org/package=psych (accessed on 11 November 2020).
- Canty, A.; Ripley, B.D. Boot: Bootstrap R (S-Plus) Functions; McMaster University: Hamilton, ON, Canada, 2020. [Google Scholar]
- Davison, A.C.; Hinkley, D.V. Bootstrap Methods and Their Applications; Cambridge University Press: Cambridge, UK, 1997; Available online: http://statwww.epfl.ch/davison/BMA/ (accessed on 29 May 2020).
- Mangiafico, S. Rcompanion: Functions to Support Extension Education Program Evaluation. 2020. Available online: https://CRAN.R-project.org/package=rcompanion (accessed on 3 November 2020).
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Muller, M.J. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Rich, J.T.; Neely, J.G.; Paniello, R.C.; Voelker, C.C.J.; Nussenbaum, B.; Wang, E. A practical guide to understanding Kaplan-Meier curves. Otolaryngol. Neck Surg. 2010, 143, 331–336. [Google Scholar] [CrossRef]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000. [Google Scholar]
- Therneau, T.M. A Package for Survival Analysis in R. 2020. Available online: https://CRAN.R-project.org/package=survival (accessed on 3 November 2021).
- Horikoshi, M.; Tang, Y. Ggfortify: Data Visualization Tools for Statistical Analysis Results. 2016. Available online: https://CRAN.R-project.org/package=ggfortify (accessed on 3 November 2020).
- Tang, Y.; Horikoshi, M.; Li, W. ggfortify: Unified Interface to Visualize Statistical Results of Popular R Packages. R J. 2016, 8, 474–485. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M. Survminer: Drawing Survival Curves Using “ggplot2”. 2016. Available online: https://CRAN.R-project.org/package=survminer (accessed on 3 November 2020).
- Langsted, A.; Nordestgaard, B.G. Nonfasting versus fasting lipid profile for cardiovascular risk prediction. Pathology 2019, 51, 131–141. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiology. 2019, 73, e285–e350. [Google Scholar] [CrossRef]
- Hoffmann, T.J.; Theusch, E.; Haldar, T.; Ranatunga, D.K.; Jorgenson, E.; Medina, M.W.; Kvale, M.N.; Kwok, P.-Y.; Schaefer, C.; Krauss, R.M.; et al. A large electronic-health-record-based genome-wide study of serum lipids. Nat. Genet. 2018, 50, 401–413. [Google Scholar] [CrossRef]
- Kanai, M.; Akiyama, M.; Takahashi, A.; Matoba, N.; Momozawa, Y.; Ikeda, M.; Iwata, N.; Ikegawa, S.; Hirata, M.; Matsuda, K.; et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 2018, 50, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Morgan, A.E.; Mooney, K.M.; Wilkinson, S.J.; Pickles, N.A.; Mc Auley, M.T. Investigating cholesterol metabolism and ageing using a systems biology approach. Proc. Nutr. Soc. 2017, 76, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.M.; Cohen, A.; Ein-Mor, E.; Stessman, J. Cholesterol, Statins, and Longevity From Age 70 to 90 Years. J. Am. Med. Dir. Assoc. 2013, 14, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Ambikairajah, A.; Walsh, E.; Cherbuin, N. Lipid profile differences during menopause: A review with meta-analysis. Menopause 2019, 26, 1327–1333. [Google Scholar] [CrossRef]
- Inaraja, V.; Thuissard, I.; Andreu-Vazquez, C.; Jodar, E. Lipid profile changes during the menopausal transition. Menopause 2020, 27, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Notelovitz, M. The effect of long-term oestrogen replacement therapy on glucose and lipid metabolism in postmenopausal women. South Afr. Med. J. 1976, 50, 2001–2003. [Google Scholar]
- Kivipelto, M.; Ngandu, T.; Laatikainen, T.; Winblad, B.; Soininen, H.; Tuomilehto, J. Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet Neurol. 2006, 5, 735–741. [Google Scholar] [CrossRef]
- Exalto, L.G.; Quesenberry, C.P.; Barnes, D.; Kivipelto, M.; Biessels, G.J.; Whitmer, R.A. Midlife risk score for the prediction of dementia four decades later. Alzheimer’s Dement. 2014, 10, 562–570. [Google Scholar] [CrossRef]
- DeCarli, C.; Villeneuve, S.; Maillard, P.; Harvey, D.; Singh, B.; Carmichael, O.; Fletcher, E.; Olichney, J.; Farias, S.; Jagust, W.; et al. Vascular Burden Score Impacts Cognition Independent of Amyloid PET and MRI Measures of Alzheimer’s Disease and Vascular Brain Injury. J. Alzheimer’s Dis. 2019, 68, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Lara, M.; Amigo, H. Association between education and blood lipid levels as income increases over a decade: A cohort study. BMC Public Health 2018, 18, 1–8. [Google Scholar] [CrossRef]
- Schilling, S.; Tzourio, C.; Soumaré, A.; Kaffashian, S.; Dartigues, J.-F.; Ancelin, M.-L.; Samieri, C.; Dufouil, C.; Debette, S. Differential associations of plasma lipids with incident dementia and dementia subtypes in the 3C Study: A longitudinal, population-based prospective cohort study. PLoS Med. 2017, 14, e1002265. [Google Scholar] [CrossRef] [PubMed]
- Picard, C.; Nilsson, N.; Labonté, A.; Auld, D.; Rosa-Neto, P.; Ashton, N.J.; Zetterberg, H.; Blennow, K.; Breitner, J.C.; Villeneuve, S.; et al. Apolipoprotein B is a novel marker for early tau pathology in Alzheimer’s disease. Alzheimer’s Dement. 2021. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PREVENT-AD | ADNI | ROSMAP | |||||
|---|---|---|---|---|---|---|---|
| Variable | N | Mean (SE) | N | Mean (SE) | N | Mean (SE) | p-Value |
| Females [%] | 306 | 69.9 (2.6) | 401 | 47.9 (2.5) | 547 | 71.1 (1.9) | <0.001 a |
| APOE-ε4 carriers [%] | 302 | 37.1 (2.8) | 401 | 56.9 (2.5) | 546 | 24.2 (1.8) | <0.001 a |
| Age [years] # | 264 | 63.33 (0.42) | 401 | 72.79 (0.35) | 547 | 88.42 (0.13) | <0.001 b |
| Statin treated [%] | 299 | 23.4 (2.5) | 360 | 51.4 (2.6) | NA | NA | <0.001 a |
| TC measurements | |||||||
| Age @ blood collection | 287 | 63.15 (0.3) | 355 | 72.63 (0.37) | NA | NA | <0.001 c |
| TC [mM] | 287 | 5.42 (0.06) | 355 | 5.09 (0.06) | NA | NA | <0.001 c |
| CSF measurements | |||||||
| Age @ CSF collection | 86 | 62.87 (0.59) | 302 | 72.19 (0.41) | NA | NA | <0.001 c |
| Aβ-42 [pg/ml] | 83 | 1160.31 (31.08) | 301 | 1053.78 (37.17) | NA | NA | 0.029 c |
| Aβ-40 [pg/ml] | 75 | 6132.53 (222.57) | NA | NA | NA | NA | NA |
| p-Tau [pg/ml] | 86 | 47.31 (1.89) | 302 | 28.03 (0.76) | NA | NA | <0.001 c |
| t-Tau [pg/ml] | 86 | 275.23 (13.87) | 302 | 291.45 (6.95) | NA | NA | 0.298 c |
| PET measurements | |||||||
| Age @ Aβ PET [years] | 98 | 67.62 (0.49) | NA | NA | NA | NA | NA |
| Aβ PET [SUVR] | 98 | 1.33 (0.04) | NA | NA | NA | NA | NA |
| Age @ TAU PET [years] | 100 | 70.79 (0.56) | NA | NA | NA | NA | NA |
| TAU PET [SUVR] | 100 | 1.07 (0.01) | NA | NA | NA | NA | NA |
| Aβ(+) | |||||||
| Age @ assessment [years] | 98 | 67.62 (0.49) | 271 | 74.22 (0.45) | 535 | 88.38 (0.13) | <0.001 b |
| Aβ(+) [%] | 98 | 18.4 (0.04) | 271 | 54.2 (3.0) | 535 | 74.6 (1.9) | <0.001 a |
| Cognition | |||||||
| Age @ assessment [years] | NA | NA | 139 | 76.62 (0.63) | 399 | 88.5 (0.15) | <0.001 c |
| CI [%] | NA | NA | 139 | 90.6 (2.5) | 399 | 76.9 (2.1) | <0.001 a |
| PREVENT-AD | ADNI | ROSMAP | ||||
|---|---|---|---|---|---|---|
| TC-PGS terms | ORs | p-Value | ORs | p-Value | ORs | p-Value |
| TC-PGS | 0.50 (0.07–3.24) | 0.25 | 0.82 (0.40–1.66) | 0.58 | 0.95 (0.66–1.37) | 0.77 |
| Statin use*TC-PGS | NA | NA | 1.68 (0.31–1.91) | 0.27 | NA | NA |
| Sex*TC-PGS | 3.64 (0.42–39.4) | 0.25 | 1.08 (0.20–1.25) | 0.87 | 0.87 (0.56-1.35) | 0.54 |
| Statin use*Sex* TC-PGS | NA | NA | 0.60 (0.05–0.53) | 0.41 | NA | NA |
| ADNI | ROSMAP | |||
|---|---|---|---|---|
| TC-PGS Terms | ORs | p-Value | ORs | p-Value |
| TC-PGS | 1.08 (0.44–2.69) | 0.86 | 0.76 (0.43–1.33) | 0.34 |
| Sex*TC-PGS | 1.18 (0.38–3.89) | 0.78 | 1.40 (0.76–2.61) | 0.28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nilsson, N.I.V.; Picard, C.; Labonté, A.; Köbe, T.; Meyer, P.-F.; Villeneuve, S.; Auld, D.; Poirier, J.; for the PREVENT-AD Research Group; Alzheimer’s Disease Neuroimaging Initiative. Association of a Total Cholesterol Polygenic Score with Cholesterol Levels and Pathological Biomarkers across the Alzheimer’s Disease Spectrum. Genes 2021, 12, 1805. https://doi.org/10.3390/genes12111805
Nilsson NIV, Picard C, Labonté A, Köbe T, Meyer P-F, Villeneuve S, Auld D, Poirier J, for the PREVENT-AD Research Group, Alzheimer’s Disease Neuroimaging Initiative. Association of a Total Cholesterol Polygenic Score with Cholesterol Levels and Pathological Biomarkers across the Alzheimer’s Disease Spectrum. Genes. 2021; 12(11):1805. https://doi.org/10.3390/genes12111805
Chicago/Turabian StyleNilsson, Nathalie I. V., Cynthia Picard, Anne Labonté, Theresa Köbe, Pierre-François Meyer, Sylvia Villeneuve, Daniel Auld, Judes Poirier, for the PREVENT-AD Research Group, and Alzheimer’s Disease Neuroimaging Initiative. 2021. "Association of a Total Cholesterol Polygenic Score with Cholesterol Levels and Pathological Biomarkers across the Alzheimer’s Disease Spectrum" Genes 12, no. 11: 1805. https://doi.org/10.3390/genes12111805
APA StyleNilsson, N. I. V., Picard, C., Labonté, A., Köbe, T., Meyer, P.-F., Villeneuve, S., Auld, D., Poirier, J., for the PREVENT-AD Research Group, & Alzheimer’s Disease Neuroimaging Initiative. (2021). Association of a Total Cholesterol Polygenic Score with Cholesterol Levels and Pathological Biomarkers across the Alzheimer’s Disease Spectrum. Genes, 12(11), 1805. https://doi.org/10.3390/genes12111805