
Translating the Role of mTOR- and RAS-Associated Signalopathies in Autism Spectrum Disorder: Models, Mechanisms and Treatment



Abstract
1. Introduction
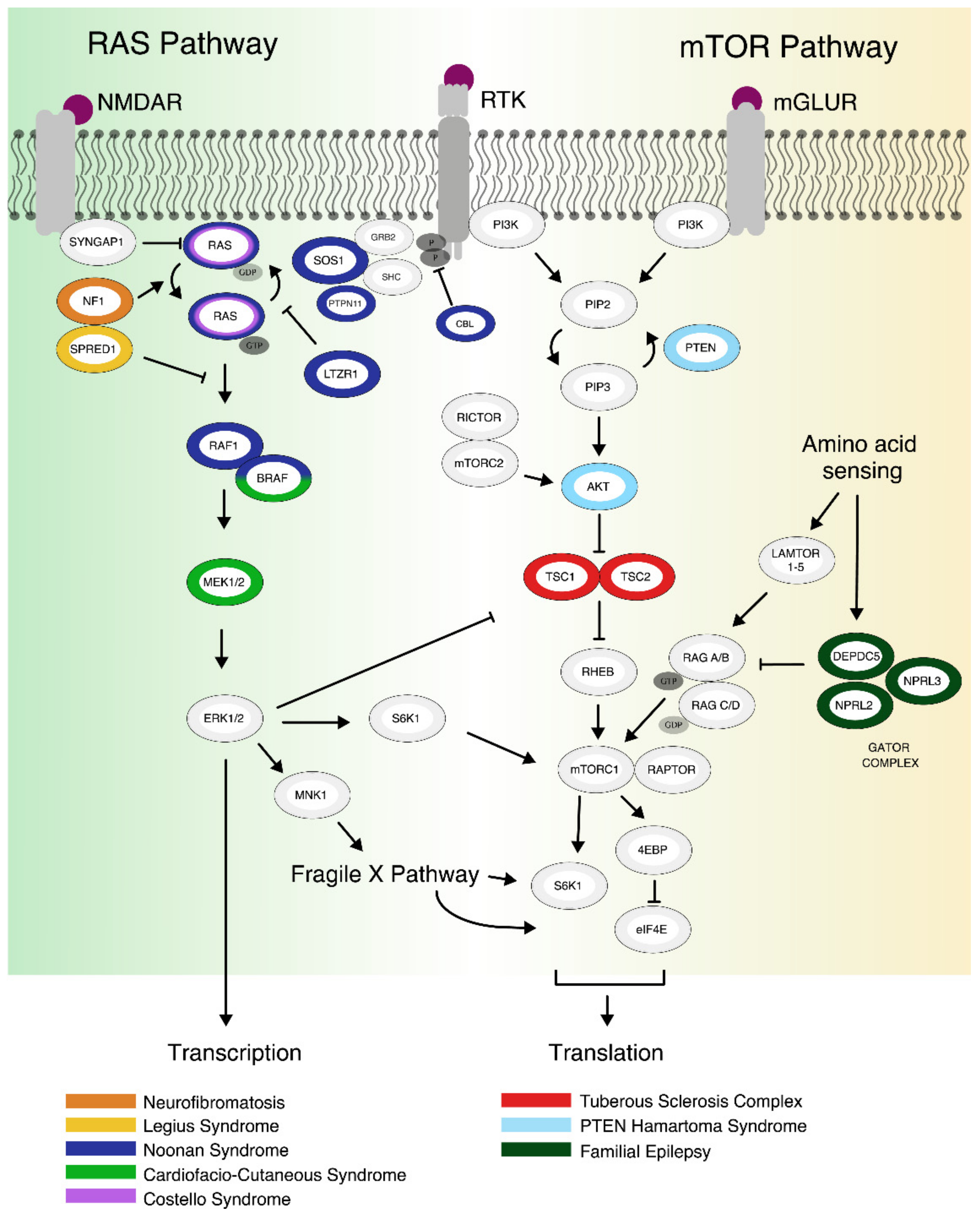
1.1. The mTOR Pathway

1.2. The RAS Pathway
2. Clinical Phenotypes and Genetics
2.1. mTORopathies
2.1.1. Tuberous Sclerosis Complex
2.1.2. PTEN Hamartoma Tumor Syndrome
2.1.3. Additional Genetic Risk Factors of the mTOR Pathway
2.2. RASopathies
2.2.1. Neurofibromatosis Type 1
2.2.2. Legius Syndrome
2.2.3. Noonan Syndrome
2.2.4. Costello Syndrome
2.2.5. CFC Syndrome
2.2.6. RAS Pathway-Related Genetic Risk Factors
3. Animal Models
3.1. Animal Models of mTORopathies
3.1.1. TSC
3.1.2. PTEN
3.1.3. mTOR Hyperactivation Models
3.2. Animal Models of RASopathies
3.2.1. NF1
3.2.2. Legius Syndrome
3.2.3. Noonan Syndrome
3.2.4. CFC Syndrome
3.2.5. Costello Syndrome
3.2.6. SYNGAP1
4. Human Cell Models
4.1. Cell Models of mTORopathies
4.1.1. TSC
4.1.2. PTEN
4.1.3. DEPDC5
4.2. Cell Models of RASopathies
4.2.1. Neurofibromatosis Type 1
4.2.2. Noonan Syndrome
4.2.3. CFC Syndrome
4.2.4. Costello Syndrome
5. Pharmacological Interventions
5.1. mTORopathies
5.2. RASopathies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| ASD | Autism Spectrum Disorder; |
| AKT | protein kinase B; |
| BRAF | V-Raf murine sarcoma viral oncogene homolog B; |
| Camp | cyclic adenosine monophosphate; |
| CBL | Casitas B-lineage Lymphoma; |
| CFCS | Cardiofacio-Cutaneous syndrome; |
| CUL3 | Cullin 3; |
| DEPDC5 | DEP domain containing 5 protein; |
| EB | embryoid bodies; |
| eIF4E | eukaryotic translation initiation factor 4E; |
| EGFR | epidermal growth factor receptor; |
| ERK 1/2 | extracellular signal-regulated kinase 1/2; |
| ESC | embryonic stem cells; |
| GABA | gamma-aminobutyric acid; |
| GAP | GTPase activating protein; |
| GATOR1 | GAP activity towards Rags 1; |
| GEF | guanine nucleotide exchange factor; |
| HCN | hyperpolarization activated cyclic nucleotide gated potassium and sodium channel; |
| ID | Intellectual Disability; |
| iPSC | induced pluripotent stem cells; |
| iNeurons | induced Neurons; |
| KO | knockout; |
| KRAS | kirsten rat sarcoma virus proto-oncogene, GTPase; |
| LAMTOR | also known as Ragulator, late endosomal/lysosomal adaptor and mitogen activated protein kinase and mechanistic target of rapamycin activator; |
| LTP | Long-term potentiation; |
| LTD | Long-term depression; |
| LZTR1 | leucine zipper–like transcriptional regulator 1; |
| MAF | minor allele frequency; |
| MAP2K1/2 or MEK 1/2 | mitogen activated protein kinase 1/2; |
| MEX3D | mex-3 RNA binding family member D; |
| MGLURs | metabotropic glutamate receptors; |
| MNK1/2 | MAPK-interacting kinase; |
| MTORC1/2 | mammalian target of rapamycin complex 1/2; |
| MWM | Morris water maze; |
| NCT | National Clinical Trial number |
| NF1 | Neurofibromatosis type 1; |
| NMDA-R | N-methyl-D-aspartic acid receptor; |
| NPC | neural progenitor cell; |
| NPRL2 | nitrogen permease regulator 2-like protein; |
| NRAS | neuroblastoma RAS viral oncogene homolog; |
| NS | Noonan Syndrome; |
| NSC | neural stem cell; |
| PI3K | Phosphoinositide 3-Kinase; |
| PTEN | Phosphatase and Tensin homolog; |
| RAF1 | Raf-1 proto-oncogene, Serine/threonine kinase; |
| Rag A/B, C/D | Ras-related GTP-binding protein A/B, C/D; |
| Raptor | regulatory-associated protein of mTOR |
| RHEB | Ras homolog enriched in brain; |
| Rictor | rapamycin-insensitive companion of mTOR; |
| RIT1 | Ras like without CAAX1; |
| RPSK | ribosomal protein S6 kinase; |
| RTK | receptor tyrosine kinases; |
| SHOC2 | SHOC2 leucine rich repeat scaffold protein 2; |
| SMAD1 | mothers against decapentaplegic homolog 1; |
| SPRED1 | sprouty-related, EVH1 domain-containing protein 1 |
| SYNGAP1 | synaptic Ras GTPase Activating Protein 1; |
| SYNGAP1-ID | SYNGAP1-related intellectual disability; |
| S6K1 | p70 S6 kinase 1; |
| TSC | Tuberous Sclerosis Complex; |
| 4E-BP1 | eukaryotic translation initiation factor 4E-binding protein. |
References
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Hultman, C.; Larsson, H.; Reichenberg, A. The Heritability of Autism Spectrum Disorder. JAMA 2017, 318, 1182–1184. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Chen, J.A.; Peñagarikano, O.; Belgard, T.G.; Swarup, V.; Geschwind, D.H. The emerging picture of autism spectrum disorder: Genetics and pathology. Annu. Rev. Pathol. 2015, 10, 111–144. [Google Scholar] [CrossRef] [PubMed]
- Borrie, S.C.; Brems, H.; Legius, E.; Bagni, C. Cognitive Dysfunctions in Intellectual Disabilities: The Contributions of the Ras-MAPK and PI3K-AKT-mTOR Pathways. Annu. Rev. Genom. Hum. Genet. 2017, 18, 115–142. [Google Scholar] [CrossRef]
- Subramanian, M.; Timmerman, C.K.; Schwartz, J.L.; Pham, D.L.; Meffert, M.K. Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci. 2015, 9, 313. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [PubMed]
- Mitra, I.; Lavillaureix, A.; Yeh, E.; Traglia, M.; Tsang, K.; Bearden, C.E.; Rauen, K.A.; Weiss, L.A. Reverse Pathway Genetic Approach Identifies Epistasis in Autism Spectrum Disorders. PLoS Genet. 2017, 13, e1006516. [Google Scholar] [CrossRef]
- Rosina, E.; Battan, B.; Siracusano, M.; Di Criscio, L.; Hollis, F.; Pacini, L.; Curatolo, P.; Bagni, C. Disruption of mTOR and MAPK pathways correlates with severity in idiopathic autism. Transl. Psychiatry 2019, 9, 50. [Google Scholar] [CrossRef]
- Gazestani, V.H.; Pramparo, T.; Nalabolu, S.; Kellman, B.P.; Murray, S.; Lopez, L.; Pierce, K.; Courchesne, E.; Lewis, N.E. A perturbed gene network containing PI3K-AKT, RAS-ERK and WNT-β-catenin pathways in leukocytes is linked to ASD genetics and symptom severity. Nat. Neurosci. 2019, 22, 1624–1634. [Google Scholar] [CrossRef]
- Li, L.; Zhao, G.-D.; Shi, Z.; Qi, L.-L.; Zhou, L.-Y.; Fu, Z.-X. The Ras/Raf/MEK/ERK signaling pathway and its role in the occurrence and development of HCC. Oncol. Lett. 2016, 12, 3045–3050. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.D.; Cruz, F. The ERK 1 and 2 pathway in the nervous system: From basic aspects to possible clinical applications in pain and visceral dysfunction. Curr. Neuropharmacol. 2007, 5, 244–252. [Google Scholar] [CrossRef]
- Kim, Y.E.; Baek, S.T. Neurodevelopmental Aspects of RASopathies. Mol. Cells 2019, 42, 441–447. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, H.-K.; Takamiya, K.; Huganir, R.L. The role of synaptic GTPase-activating protein in neuronal development and synaptic plasticity. J. Neurosci. 2003, 23, 1119–1124. [Google Scholar] [CrossRef]
- Steklov, M.; Pandolfi, S.; Baietti, M.F.; Batiuk, A.; Carai, P.; Najm, P.; Zhang, M.; Jang, H.; Renzi, F.; Cai, Y.; et al. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 2018, 362, 1177–1182. [Google Scholar] [CrossRef]
- Bigenzahn, J.W.; Collu, G.M.; Kartnig, F.; Pieraks, M.; Vladimer, G.I.; Heinz, L.X.; Sedlyarov, V.; Schischlik, F.; Fauster, A.; Rebsamen, M.; et al. LZTR1 is a regulator of RAS ubiquitination and signaling. Science 2018, 362, 1171–1177. [Google Scholar] [CrossRef]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Satterstrom, F.K.; Kosmicki, J.A.; Wang, J.; Breen, M.S.; de Rubeis, S.; An, J.-Y.; Peng, M.; Collins, R.; Grove, J.; Klei, L.; et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 2020, 180, 568–584.e23. [Google Scholar] [CrossRef]
- Iossifov, I.; O’Roak, B.J.; Sanders, S.J.; Ronemus, M.; Krumm, N.; Levy, D.; Stessman, H.A.; Witherspoon, K.T.; Vives, L.; Patterson, K.E.; et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014, 515, 216–221. [Google Scholar] [CrossRef]
- Rylaarsdam, L.; Guemez-Gamboa, A. Genetic Causes and Modifiers of Autism Spectrum Disorder. Front. Cell. Neurosci. 2019, 13, 385. [Google Scholar] [CrossRef]
- Jesus-Ribeiro, J.; Pires, L.M.; Melo, J.D.; Ribeiro, I.P.; Rebelo, O.; Sales, F.; Freire, A.; Melo, J.B. Genomic and Epigenetic Advances in Focal Cortical Dysplasia Types I and II: A Scoping Review. Front. Neurosci. 2020, 14, 580357. [Google Scholar] [CrossRef] [PubMed]
- Jeste, S.S.; Varcin, K.J.; Hellemann, G.S.; Gulsrud, A.C.; Bhatt, R.; Kasari, C.; Wu, J.Y.; Sahin, M.; Nelson, C.A. Symptom profiles of autism spectrum disorder in tuberous sclerosis complex. Neurology 2016, 87, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Henske, E.P.; Jóźwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Richards, C.; Jones, C.; Groves, L.; Moss, J.; Oliver, C. Prevalence of autism spectrum disorder phenomenology in genetic disorders: A systematic review and meta-analysis. Lancet Psychiatry 2015, 2, 909–916. [Google Scholar] [CrossRef]
- Jones, A.C.; Daniells, C.E.; Snell, R.G.; Tachataki, M.; Idziaszczyk, S.A.; Krawczak, M.; Sampson, J.R.; Cheadle, J.P. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum. Mol. Genet. 1997, 6, 2155–2161. [Google Scholar] [CrossRef]
- Betancur, C. Etiological heterogeneity in autism spectrum disorders: More than 100 genetic and genomic disorders and still counting. Brain Res. 2011, 1380, 42–77. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Yehia, L.; Keel, E.; Eng, C. The Clinical Spectrum of PTEN Mutations. Annu. Rev. Med. 2020, 71, 103–116. [Google Scholar] [CrossRef]
- Nelen, M.R.; Kremer, H.; Konings, I.B.; Schoute, F.; van Essen, A.J.; Koch, R.; Woods, C.G.; Fryns, J.P.; Hamel, B.; Hoefsloot, L.H.; et al. Novel PTEN mutations in patients with Cowden disease: Absence of clear genotype-phenotype correlations. Eur. J. Hum. Genet. 1999, 7, 267–273. [Google Scholar] [CrossRef]
- Zhou, X.-P.; Waite, K.A.; Pilarski, R.; Hampel, H.; Fernandez, M.J.; Bos, C.; Dasouki, M.; Feldman, G.L.; Greenberg, L.A.; Ivanovich, J.; et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2003, 73, 404–411. [Google Scholar] [CrossRef]
- Busch, R.M.; Srivastava, S.; Hogue, O.; Frazier, T.W.; Klaas, P.; Hardan, A.; Martinez-Agosto, J.A.; Sahin, M.; Eng, C. Neurobehavioral phenotype of autism spectrum disorder associated with germline heterozygous mutations in PTEN. Transl. Psychiatry 2019, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.S.; Berkovic, S.F.; Cossette, P.; Delanty, N.; Dlugos, D.; Eichler, E.E.; Epstein, M.P.; Glauser, T.; Goldstein, D.B.; Han, Y.; et al. De novo mutations in epileptic encephalopathies. Nature 2013, 501, 217–221. [Google Scholar] [CrossRef]
- Reijnders, M.R.F.; Kousi, M.; van Woerden, G.M.; Klein, M.; Bralten, J.; Mancini, G.M.S.; van Essen, T.; Proietti-Onori, M.; Smeets, E.E.J.; van Gastel, M.; et al. Variation in a range of mTOR-related genes associates with intracranial volume and intellectual disability. Nat. Commun. 2017, 8, 1052. [Google Scholar] [CrossRef]
- Mirzaa, G.M.; Campbell, C.D.; Solovieff, N.; Goold, C.; Jansen, L.A.; Menon, S.; Timms, A.E.; Conti, V.; Biag, J.D.; Adams, C.; et al. Association of MTOR Mutations With Developmental Brain Disorders, Including Megalencephaly, Focal Cortical Dysplasia, and Pigmentary Mosaicism. JAMA Neurol. 2016, 73, 836–845. [Google Scholar] [CrossRef]
- Baldassari, S.; Picard, F.; Verbeek, N.E.; van Kempen, M.; Brilstra, E.H.; Lesca, G.; Conti, V.; Guerrini, R.; Bisulli, F.; Licchetta, L.; et al. The landscape of epilepsy-related GATOR1 variants. Genet. Med. 2019, 21, 398–408. [Google Scholar] [CrossRef]
- Ribierre, T.; Deleuze, C.; Bacq, A.; Baldassari, S.; Marsan, E.; Chipaux, M.; Muraca, G.; Roussel, D.; Navarro, V.; Leguern, E.; et al. Second-hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia-associated epilepsy. J. Clin. Investig. 2018, 128, 2452–2458. [Google Scholar] [CrossRef]
- Li, S.; Balmain, A.; Counter, C.M. A model for RAS mutation patterns in cancers: Finding the sweet spot. Nat. Rev. Cancer 2018, 18, 767–777. [Google Scholar] [CrossRef]
- Rauen, K.A. The RASopathies. Annu. Rev. Genom. Hum. Genet. 2013, 14, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.C.; Lucas, J.; Babcock, M.A.; Gutmann, D.H.; Korf, B.; Maria, B.L. Neurofibromatosis type 1 revisited. Pediatrics 2009, 123, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Vogel, A.C.; Gutmann, D.H.; Morris, S.M. Neurodevelopmental disorders in children with neurofibromatosis type 1. Dev. Med. Child Neurol. 2017, 59, 1112–1116. [Google Scholar] [CrossRef]
- Eijk, S.; Mous, S.E.; Dieleman, G.C.; Dierckx, B.; Rietman, A.B.; de Nijs, P.F.A.; ten Hoopen, L.W.; van Minkelen, R.; Elgersma, Y.; Catsman-Berrevoets, C.E.; et al. Autism Spectrum Disorder in an Unselected Cohort of Children with Neurofibromatosis Type 1 (NF1). J. Autism Dev. Disord. 2018, 48, 2278–2285. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Plasschaert, E.; Descheemaeker, M.-J.; Huson, S.; Borghgraef, M.; Vogels, A.; Evans, D.G.; Legius, E.; Green, J. Autism spectrum disorder profile in neurofibromatosis type I. J. Autism Dev. Disord. 2015, 45, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Stowe, I.B.; Mercado, E.L.; Stowe, T.R.; Bell, E.L.; Oses-Prieto, J.A.; Hernández, H.; Burlingame, A.L.; McCormick, F. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012, 26, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Chmara, M.; Sahbatou, M.; Denayer, E.; Taniguchi, K.; Kato, R.; Somers, R.; Messiaen, L.; de Schepper, S.; Fryns, J.-P.; et al. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat. Genet. 2007, 39, 1120–1126. [Google Scholar] [CrossRef] [PubMed]
- Brems, H.; Pasmant, E.; van Minkelen, R.; Wimmer, K.; Upadhyaya, M.; Legius, E.; Messiaen, L. Review and update of SPRED1 mutations causing Legius syndrome. Hum. Mutat. 2012, 33, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, M.; Mehler, E.L.; Goldberg, R.; Zampino, G.; Brunner, H.G.; Kremer, H.; van der Burgt, I.; Crosby, A.H.; Ion, A.; Jeffery, S.; et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet. 2001, 29, 465–468. [Google Scholar] [CrossRef]
- Tartaglia, M.; Pennacchio, L.A.; Zhao, C.; Yadav, K.K.; Fodale, V.; Sarkozy, A.; Pandit, B.; Oishi, K.; Martinelli, S.; Schackwitz, W.; et al. Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 2007, 39, 75–79. [Google Scholar] [CrossRef]
- Sewduth, R.N.; Pandolfi, S.; Steklov, M.; Sheryazdanova, A.; Zhao, P.; Criem, N.; Baietti, M.F.; Lechat, B.; Quarck, R.; Impens, F.; et al. The Noonan Syndrome Gene Lztr1 Controls Cardiovascular Function by Regulating Vesicular Trafficking. Circ. Res. 2020, 126, 1379–1393. [Google Scholar] [CrossRef]
- Tidyman, W.E.; Rauen, K.A. Expansion of the RASopathies. Curr. Genet. Med. Rep. 2016, 4, 57–64. [Google Scholar] [CrossRef]
- Roberts, A.E.; Allanson, J.E.; Tartaglia, M.; Gelb, B.D. Noonan syndrome. Lancet 2013, 381, 333–342. [Google Scholar] [CrossRef]
- Gripp, K.W.; Hopkins, E.; Sol-Church, K.; Stabley, D.L.; Axelrad, M.E.; Doyle, D.; Dobyns, W.B.; Hudson, C.; Johnson, J.; Tenconi, R.; et al. Phenotypic analysis of individuals with Costello syndrome due to HRAS p.G13C. Am. J. Med. Genet. A 2011, 155, 706–716. [Google Scholar] [CrossRef]
- Gripp, K.W.; Lin, A.E. Costello syndrome: A Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet. Med. 2012, 14, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Adviento, B.; Corbin, I.L.; Widjaja, F.; Desachy, G.; Enrique, N.; Rosser, T.; Risi, S.; Marco, E.J.; Hendren, R.L.; Bearden, C.E.; et al. Autism traits in the RASopathies. J. Med. Genet. 2014, 51, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Alfieri, P.; Piccini, G.; Caciolo, C.; Perrino, F.; Gambardella, M.L.; Mallardi, M.; Cesarini, L.; Leoni, C.; Leone, D.; Fossati, C.; et al. Behavioral profile in RASopathies. Am. J. Med. Genet. A 2014, 164, 934–942. [Google Scholar] [CrossRef]
- Pierpont, M.E.M.; Magoulas, P.L.; Adi, S.; Kavamura, M.I.; Neri, G.; Noonan, J.; Pierpont, E.I.; Reinker, K.; Roberts, A.E.; Shankar, S.; et al. Cardio-facio-cutaneous syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics 2014, 134, e1149-62. [Google Scholar] [CrossRef]
- Roberts, A.; Allanson, J.; Jadico, S.K.; Kavamura, M.I.; Noonan, J.; Opitz, J.M.; Young, T.; Neri, G. The cardiofaciocutaneous syndrome. J. Med. Genet. 2006, 43, 833–842. [Google Scholar] [CrossRef]
- Holder, J.L.; Hamdan, F.F.; Michaud, J.L. SYNGAP1-Related Intellectual Disability; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; GeneReviews; University of Washington: Seattle, WA, USA, 1993; pp. 1993–2021. [Google Scholar]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’Dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A.; et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.L.M.; Gilissen, C.; Veltman, J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016, 17, 9–18. [Google Scholar] [CrossRef]
- Wang, T.; Hoekzema, K.; Vecchio, D.; Wu, H.; Sulovari, A.; Coe, B.P.; Gillentine, M.A.; Wilfert, A.B.; Perez-Jurado, L.A.; Kvarnung, M.; et al. Large-scale targeted sequencing identifies risk genes for neurodevelopmental disorders. Nat. Commun. 2020, 11, 4932. [Google Scholar] [CrossRef] [PubMed]
- Potter, W.B.; Basu, T.; O’Riordan, K.J.; Kirchner, A.; Rutecki, P.; Burger, C.; Roopra, A. Reduced juvenile long-term depression in tuberous sclerosis complex is mitigated in adults by compensatory recruitment of mGluR5 and Erk signaling. PLoS Biol. 2013, 11, e1001627. [Google Scholar] [CrossRef]
- Reith, R.M.; McKenna, J.; Wu, H.; Hashmi, S.S.; Cho, S.-H.; Dash, P.K.; Gambello, M.J. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiol. Dis. 2013, 51, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Bateup, H.S.; Johnson, C.A.; Denefrio, C.L.; Saulnier, J.L.; Kornacker, K.; Sabatini, B.L. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013, 78, 510–522. [Google Scholar] [CrossRef]
- Abs, E.; Goorden, S.M.I.; Schreiber, J.; Overwater, I.E.; Hoogeveen-Westerveld, M.; Bruinsma, C.F.; Aganović, E.; Borgesius, N.Z.; Nellist, M.; Elgersma, Y. TORC1-dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann. Neurol. 2013, 74, 569–579. [Google Scholar] [CrossRef]
- Ehninger, D.; Han, S.; Shilyansky, C.; Zhou, Y.; Li, W.; Kwiatkowski, D.J.; Ramesh, V.; Silva, A.J. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat. Med. 2008, 14, 843–848. [Google Scholar] [CrossRef]
- Meikle, L.; Talos, D.M.; Onda, H.; Pollizzi, K.; Rotenberg, A.; Sahin, M.; Jensen, F.E.; Kwiatkowski, D.J. A mouse model of tuberous sclerosis: Neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J. Neurosci. 2007, 27, 5546–5558. [Google Scholar] [CrossRef]
- Sato, A.; Kasai, S.; Kobayashi, T.; Takamatsu, Y.; Hino, O.; Ikeda, K.; Mizuguchi, M. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat. Commun. 2012, 3, 1292. [Google Scholar] [CrossRef] [PubMed]
- Goorden, S.M.I.; van Woerden, G.M.; van der Weerd, L.; Cheadle, J.P.; Elgersma, Y. Cognitive deficits in Tsc1+/- mice in the absence of cerebral lesions and seizures. Ann. Neurol. 2007, 62, 648–655. [Google Scholar] [CrossRef]
- Carson, R.P.; van Nielen, D.L.; Winzenburger, P.A.; Ess, K.C. Neuronal and glia abnormalities in Tsc1-deficient forebrain and partial rescue by rapamycin. Neurobiol. Dis. 2012, 45, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Carson, R.P.; Kelm, N.D.; West, K.L.; Does, M.D.; Fu, C.; Weaver, G.; McBrier, E.; Parker, B.; Grier, M.D.; Ess, K.C. Hypomyelination following deletion of Tsc2 in oligodendrocyte precursors. Ann. Clin. Transl. Neurol. 2015, 2, 1041–1054. [Google Scholar] [CrossRef]
- Crowell, B.; Lee, G.H.; Nikolaeva, I.; Dal Pozzo, V.; D’Arcangelo, G. Complex Neurological Phenotype in Mutant Mice Lacking Tsc2 in Excitatory Neurons of the Developing Forebrain(123). Eneuro 2015, 2, ENEURO.0046-15.2015. [Google Scholar] [CrossRef] [PubMed]
- Backman, S.A.; Stambolic, V.; Suzuki, A.; Haight, J.; Elia, A.; Pretorius, J.; Tsao, M.S.; Shannon, P.; Bolon, B.; Ivy, G.O.; et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat. Genet. 2001, 29, 396–403. [Google Scholar] [CrossRef]
- Fraser, M.M.; Bayazitov, I.T.; Zakharenko, S.S.; Baker, S.J. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience 2008, 151, 476–488. [Google Scholar] [CrossRef]
- Zhou, J.; Blundell, J.; Ogawa, S.; Kwon, C.-H.; Zhang, W.; Sinton, C.; Powell, C.M.; Parada, L.F. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J. Neurosci. 2009, 29, 1773–1783. [Google Scholar] [CrossRef]
- Amiri, A.; Cho, W.; Zhou, J.; Birnbaum, S.G.; Sinton, C.M.; McKay, R.M.; Parada, L.F. Pten deletion in adult hippocampal neural stem/progenitor cells causes cellular abnormalities and alters neurogenesis. J. Neurosci. 2012, 32, 5880–5890. [Google Scholar] [CrossRef] [PubMed]
- Luikart, B.W.; Schnell, E.; Washburn, E.K.; Bensen, A.L.; Tovar, K.R.; Westbrook, G.L. Pten knockdown in vivo increases excitatory drive onto dentate granule cells. J. Neurosci. 2011, 31, 4345–4354. [Google Scholar] [CrossRef]
- Haws, M.E.; Jaramillo, T.C.; Espinosa, F.; Widman, A.J.; Stuber, G.D.; Sparta, D.R.; Tye, K.M.; Russo, S.J.; Parada, L.F.; Stavarache, M.; et al. PTEN knockdown alters dendritic spine/protrusion morphology, not density. J. Comp. Neurol. 2014, 522, 1171–1190. [Google Scholar] [CrossRef] [PubMed]
- Yehia, L.; Eng, C. 65 Years of the double helix: One gene, many endocrine and metabolic syndromes: PTEN-opathies and precision medicine. Endocr. Relat. Cancer 2018, 25, T121–T140. [Google Scholar] [CrossRef]
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of dendritic arborization by the phosphoinositide-3′-kinase-Akt-mammalian target of rapamycin pathway. J. Neurosci. 2005, 25, 11300–11312. [Google Scholar] [CrossRef]
- Tschopp, O.; Yang, Z.-Z.; Brodbeck, D.; Dummler, B.A.; Hemmings-Mieszczak, M.; Watanabe, T.; Michaelis, T.; Frahm, J.; Hemmings, B.A. Essential role of protein kinase B gamma (PKB gamma/Akt3) in postnatal brain development but not in glucose homeostasis. Development 2005, 132, 2943–2954. [Google Scholar] [CrossRef]
- Easton, R.M.; Cho, H.; Roovers, K.; Shineman, D.W.; Mizrahi, M.; Forman, M.S.; Lee, V.M.-Y.; Szabolcs, M.; de Jong, R.; Oltersdorf, T.; et al. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol. Cell. Biol. 2005, 25, 1869–1878. [Google Scholar] [CrossRef]
- Santini, E.; Huynh, T.N.; MacAskill, A.F.; Carter, A.G.; Pierre, P.; Ruggero, D.; Kaphzan, H.; Klann, E. Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 2013, 493, 411–415. [Google Scholar] [CrossRef]
- Lin, T.V.; Hsieh, L.; Kimura, T.; Malone, T.J.; Bordey, A. Normalizing translation through 4E-BP prevents mTOR-driven cortical mislamination and ameliorates aberrant neuron integration. Proc. Natl. Acad. Sci. USA 2016, 113, 11330–11335. [Google Scholar] [CrossRef]
- Banko, J.L.; Poulin, F.; Hou, L.; DeMaria, C.T.; Sonenberg, N.; Klann, E. The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J. Neurosci. 2005, 25, 9581–9590. [Google Scholar] [CrossRef]
- Koehl, M.; Ladevèze, E.; Catania, C.; Cota, D.; Abrous, D.N. Inhibition of mTOR signaling by genetic removal of p70 S6 kinase 1 increases anxiety-like behavior in mice. Transl. Psychiatry 2021, 11, 165. [Google Scholar] [CrossRef]
- Ruvinsky, I.; Sharon, N.; Lerer, T.; Cohen, H.; Stolovich-Rain, M.; Nir, T.; Dor, Y.; Zisman, P.; Meyuhas, O. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005, 19, 2199–2211. [Google Scholar] [CrossRef] [PubMed]
- Antion, M.D.; Merhav, M.; Hoeffer, C.A.; Reis, G.; Kozma, S.C.; Thomas, G.; Schuman, E.M.; Rosenblum, K.; Klann, E. Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn. Mem. 2008, 15, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Cao, R.; Jafarnejad, S.M.; Prager-Khoutorsky, M.; Giannakas, N.; Kaminari, A.; Fragkouli, A.; Nader, K.; Price, T.J.; et al. Pharmacogenetic inhibition of eIF4E-dependent Mmp9 mRNA translation reverses fragile X syndrome-like phenotypes. Cell Rep. 2014, 9, 1742–1755. [Google Scholar] [CrossRef]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Stevens, D.M.; Thoreen, C.C.; Burds, A.A.; Kalaany, N.Y.; Moffat, J.; Brown, M.; Fitzgerald, K.J.; Sabatini, D.M. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 2006, 11, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Shiota, C.; Woo, J.-T.; Lindner, J.; Shelton, K.D.; Magnuson, M.A. Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev. Cell 2006, 11, 583–589. [Google Scholar] [CrossRef]
- Ka, M.; Condorelli, G.; Woodgett, J.R.; Kim, W.-Y. mTOR regulates brain morphogenesis by mediating GSK3 signaling. Development 2014, 141, 4076–4086. [Google Scholar] [CrossRef]
- Ohne, Y.; Takahara, T.; Hatakeyama, R.; Matsuzaki, T.; Noda, M.; Mizushima, N.; Maeda, T. Isolation of hyperactive mutants of mammalian target of rapamycin. J. Biol. Chem. 2008, 283, 31861–31870. [Google Scholar] [CrossRef]
- Kassai, H.; Sugaya, Y.; Noda, S.; Nakao, K.; Maeda, T.; Kano, M.; Aiba, A. Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep. 2014, 7, 1626–1639. [Google Scholar] [CrossRef]
- Marsan, E.; Ishida, S.; Schramm, A.; Weckhuysen, S.; Muraca, G.; Lecas, S.; Liang, N.; Treins, C.; Pende, M.; Roussel, D.; et al. Depdc5 knockout rat: A novel model of mTORopathy. Neurobiol. Dis. 2016, 89, 180–189. [Google Scholar] [CrossRef]
- Yuskaitis, C.J.; Jones, B.M.; Wolfson, R.L.; Super, C.E.; Dhamne, S.C.; Rotenberg, A.; Sabatini, D.M.; Sahin, M.; Poduri, A. A mouse model of DEPDC5-related epilepsy: Neuronal loss of Depdc5 causes dysplastic and ectopic neurons, increased mTOR signaling, and seizure susceptibility. Neurobiol. Dis. 2018, 111, 91–101. [Google Scholar] [CrossRef]
- Hu, S.; Knowlton, R.C.; Watson, B.O.; Glanowska, K.M.; Murphy, G.G.; Parent, J.M.; Wang, Y. Somatic Depdc5 deletion recapitulates electroclinical features of human focal cortical dysplasia type IIA. Ann. Neurol. 2018, 84, 140–146. [Google Scholar] [CrossRef]
- Iffland, P.H.; Baybis, M.; Barnes, A.E.; Leventer, R.J.; Lockhart, P.J.; Crino, P.B. DEPDC5 and NPRL3 modulate cell size, filopodial outgrowth, and localization of mTOR in neural progenitor cells and neurons. Neurobiol. Dis. 2018, 114, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Goorden, S.M.I.; Abs, E.; Bruinsma, C.F.; Riemslagh, F.W.; van Woerden, G.M.; Elgersma, Y. Intact neuronal function in Rheb1 mutant mice: Implications for TORC1-based treatments. Hum. Mol. Genet. 2015, 24, 3390–3398. [Google Scholar] [CrossRef] [PubMed]
- Park, S.M.; Lim, J.S.; Ramakrishina, S.; Kim, S.H.; Kim, W.K.; Lee, J.; Kang, H.-C.; Reiter, J.F.; Kim, D.S.; Kim, H.H.; et al. Brain Somatic Mutations in MTOR Disrupt Neuronal Ciliogenesis, Leading to Focal Cortical Dyslamination. Neuron 2018, 99, 83–97.e7. [Google Scholar] [CrossRef] [PubMed]
- Hanai, S.; Sukigara, S.; Dai, H.; Owa, T.; Horike, S.-I.; Otsuki, T.; Saito, T.; Nakagawa, E.; Ikegaya, N.; Kaido, T.; et al. Pathologic Active mTOR Mutation in Brain Malformation with Intractable Epilepsy Leads to Cell-Autonomous Migration Delay. Am. J. Pathol. 2017, 187, 1177–1185. [Google Scholar] [CrossRef]
- Wang, H.-F.; Shih, Y.-T.; Chen, C.-Y.; Chao, H.-W.; Lee, M.-J.; Hsueh, Y.-P. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Investig. 2011, 121, 4820–4837. [Google Scholar] [CrossRef]
- Omrani, A.; van der Vaart, T.; Mientjes, E.; van Woerden, G.M.; Hojjati, M.R.; Li, K.W.; Gutmann, D.H.; Levelt, C.N.; Smit, A.B.; Silva, A.J.; et al. HCN channels are a novel therapeutic target for cognitive dysfunction in Neurofibromatosis type 1. Mol. Psychiatry 2015, 20, 1311–1321. [Google Scholar] [CrossRef]
- Shilyansky, C.; Karlsgodt, K.H.; Cummings, D.M.; Sidiropoulou, K.; Hardt, M.; James, A.S.; Ehninger, D.; Bearden, C.E.; Poirazi, P.; Jentsch, J.D.; et al. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc. Natl. Acad. Sci. USA 2010, 107, 13141–13146. [Google Scholar] [CrossRef]
- Costa, R.M.; Federov, N.B.; Kogan, J.H.; Murphy, G.G.; Stern, J.; Ohno, M.; Kucherlapati, R.; Jacks, T.; Silva, A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 2002, 415, 526–530. [Google Scholar] [CrossRef]
- Cui, Y.; Costa, R.M.; Murphy, G.G.; Elgersma, Y.; Zhu, Y.; Gutmann, D.H.; Parada, L.F.; Mody, I.; Silva, A.J. Neurofibromin Regulation of ERK Signaling Modulates GABA Release and Learning. Cell 2008, 135, 549–560. [Google Scholar] [CrossRef]
- Denayer, E.; Ahmed, T.; Brems, H.; van Woerden, G.; Borgesius, N.Z.; Callaerts-Vegh, Z.; Yoshimura, A.; Hartmann, D.; Elgersma, Y.; D’Hooge, R.; et al. Spred1 is required for synaptic plasticity and hippocampus-dependent learning. J. Neurosci. 2008, 28, 14443–14449. [Google Scholar] [CrossRef]
- Borrie, S.C.; Horner, A.E.; Yoshimura, A.; Legius, E.; Kopanitsa, M.V.; Brems, H. Impaired instrumental learning in Spred1-/- mice, a model for a rare RASopathy. Genes Brain Behav. 2021, 20, e12727. [Google Scholar] [CrossRef] [PubMed]
- Borrie, S.C.; Plasschaert, E.; Callaerts-Vegh, Z.; Yoshimura, A.; D’Hooge, R.; Elgersma, Y.; Kushner, S.A.; Legius, E.; Brems, H. MEK inhibition ameliorates social behavior phenotypes in a Spred1 knockout mouse model for RASopathy disorders. Mol. Autism 2021, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Ehninger, D.; Zhou, M.; Oh, J.-Y.; Kang, M.; Kwak, C.; Ryu, H.-H.; Butz, D.; Araki, T.; Cai, Y.; et al. Mechanism and treatment for learning and memory deficits in mouse models of Noonan syndrome. Nat. Neurosci. 2014, 17, 1736–1743. [Google Scholar] [CrossRef]
- Kusakari, S.; Saitow, F.; Ago, Y.; Shibasaki, K.; Sato-Hashimoto, M.; Matsuzaki, Y.; Kotani, T.; Murata, Y.; Hirai, H.; Matsuda, T.; et al. Shp2 in forebrain neurons regulates synaptic plasticity, locomotion, and memory formation in mice. Mol. Cell. Biol. 2015, 35, 1557–1572. [Google Scholar] [CrossRef]
- Zheng, H.; Yu, W.-M.; Waclaw, R.R.; Kontaridis, M.I.; Neel, B.G.; Qu, C.-K. Gain-of-function mutations in the gene encoding the tyrosine phosphatase SHP2 induce hydrocephalus in a catalytically dependent manner. Sci. Signal. 2018, 11, eaao1591. [Google Scholar] [CrossRef]
- Holter, M.C.; Hewitt, L.T.; Koebele, S.V.; Judd, J.M.; Xing, L.; Bimonte-Nelson, H.A.; Conrad, C.D.; Araki, T.; Neel, B.G.; Snider, W.D.; et al. The Noonan Syndrome-linked Raf1L613V mutation drives increased glial number in the mouse cortex and enhanced learning. PLoS Genet. 2019, 15, e1008108. [Google Scholar] [CrossRef]
- Inoue, S.-I.; Moriya, M.; Watanabe, Y.; Miyagawa-Tomita, S.; Niihori, T.; Oba, D.; Ono, M.; Kure, S.; Ogura, T.; Matsubara, Y.; et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum. Mol. Genet. 2014, 23, 6553–6566. [Google Scholar] [CrossRef]
- Moriya, M.; Inoue, S.-I.; Miyagawa-Tomita, S.; Nakashima, Y.; Oba, D.; Niihori, T.; Hashi, M.; Ohnishi, H.; Kure, S.; Matsubara, Y.; et al. Adult mice expressing a Braf Q241R mutation on an ICR/CD-1 background exhibit a cardio-facio-cutaneous syndrome phenotype. Hum. Mol. Genet. 2015, 24, 7349–7360. [Google Scholar] [CrossRef]
- Urosevic, J.; Sauzeau, V.; Soto-Montenegro, M.L.; Reig, S.; Desco, M.; Wright, E.M.B.; Cañamero, M.; Mulero, F.; Ortega, S.; Bustelo, X.R.; et al. Constitutive activation of B-Raf in the mouse germ line provides a model for human cardio-facio-cutaneous syndrome. Proc. Natl. Acad. Sci. USA 2011, 108, 5015–5020. [Google Scholar] [CrossRef]
- Koh, H.Y.; Kim, S.H.; Jang, J.; Kim, H.; Han, S.; Lim, J.S.; Son, G.; Choi, J.; Park, B.O.; Heo, W.D.; et al. BRAF somatic mutation contributes to intrinsic epileptogenicity in pediatric brain tumors. Nat. Med. 2018, 24, 1662–1668. [Google Scholar] [CrossRef]
- Chen, A.P.; Ohno, M.; Giese, K.P.; Kühn, R.; Chen, R.L.; Silva, A.J. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J. Neurosci. Res. 2006, 83, 28–38. [Google Scholar] [CrossRef]
- Papale, A.; d’Isa, R.; Menna, E.; Cerovic, M.; Solari, N.; Hardingham, N.; Cambiaghi, M.; Cursi, M.; Barbacid, M.; Leocani, L.; et al. Severe Intellectual Disability and Enhanced Gamma-Aminobutyric Acidergic Synaptogenesis in a Novel Model of Rare RASopathies. Biol. Psychiatry 2017, 81, 179–192. [Google Scholar] [CrossRef]
- Kelleher, R.J.; Govindarajan, A.; Jung, H.-Y.; Kang, H.; Tonegawa, S. Translational Control by MAPK Signaling in Long-Term Synaptic Plasticity and Memory. Cell 2004, 116, 467–479. [Google Scholar] [CrossRef]
- Aoidi, R.; Houde, N.; Landry-Truchon, K.; Holter, M.; Jacquet, K.; Charron, L.; Krishnaswami, S.R.; Yu, B.D.; Rauen, K.A.; Bisson, N.; et al. Mek1Y130C mice recapitulate aspects of human cardio-facio-cutaneous syndrome. Dis. Model. Mech. 2018, 11, dmm031278. [Google Scholar] [CrossRef]
- Holter, M.C.; Hewitt, L.T.; Nishimura, K.J.; Knowles, S.J.; Bjorklund, G.R.; Shah, S.; Fry, N.R.; Rees, K.P.; Gupta, T.A.; Daniels, C.W.; et al. Hyperactive MEK1 Signaling in Cortical GABAergic Neurons Promotes Embryonic Parvalbumin Neuron Loss and Defects in Behavioral Inhibition. Cereb. Cortex 2021, 31, 3064–3081. [Google Scholar] [CrossRef]
- Viosca, J.; Schuhmacher, A.J.; Guerra, C.; Barco, A. Germline expression of H-Ras(G12V) causes neurological deficits associated to Costello syndrome. Genes Brain Behav. 2009, 8, 60–71. [Google Scholar] [CrossRef]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhöhl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic activation of Ras in neurons promotes hypertrophy and protects from lesion-induced degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef]
- Rooney, G.E.; Goodwin, A.F.; Depeille, P.; Sharir, A.; Schofield, C.M.; Yeh, E.; Roose, J.P.; Klein, O.D.; Rauen, K.A.; Weiss, L.A.; et al. Human iPS Cell-Derived Neurons Uncover the Impact of Increased Ras Signaling in Costello Syndrome. J. Neurosci. 2016, 36, 142–152. [Google Scholar] [CrossRef]
- Krencik, R.; Hokanson, K.C.; Narayan, A.R.; Dvornik, J.; Rooney, G.E.; Rauen, K.A.; Weiss, L.A.; Rowitch, D.H.; Ullian, E.M. Dysregulation of astrocyte extracellular signaling in Costello syndrome. Sci. Transl. Med. 2015, 7, 286ra66. [Google Scholar] [CrossRef]
- Kushner, S.A.; Elgersma, Y.; Murphy, G.G.; Jaarsma, D.; van Woerden, G.M.; Hojjati, M.R.; Cui, Y.; LeBoutillier, J.C.; Marrone, D.F.; Choi, E.S.; et al. Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J. Neurosci. 2005, 25, 9721–9734. [Google Scholar] [CrossRef]
- Clement, J.P.; Aceti, M.; Creson, T.K.; Ozkan, E.D.; Shi, Y.; Reish, N.J.; Almonte, A.G.; Miller, B.H.; Wiltgen, B.J.; Miller, C.A.; et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 2012, 151, 709–723. [Google Scholar] [CrossRef]
- Barnes, S.A.; Wijetunge, L.S.; Jackson, A.D.; Katsanevaki, D.; Osterweil, E.K.; Komiyama, N.H.; Grant, S.G.N.; Bear, M.F.; Nägerl, U.V.; Kind, P.C.; et al. Convergence of Hippocampal Pathophysiology in Syngap+/- and Fmr1-/y Mice. J. Neurosci. 2015, 35, 15073–15081. [Google Scholar] [CrossRef]
- Komiyama, N.H.; Watabe, A.M.; Carlisle, H.J.; Porter, K.; Charlesworth, P.; Monti, J.; Strathdee, D.J.C.; O’Carroll, C.M.; Martin, S.J.; Morris, R.G.M.; et al. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J. Neurosci. 2002, 22, 9721–9732. [Google Scholar] [CrossRef]
- Clement, J.P.; Ozkan, E.D.; Aceti, M.; Miller, C.A.; Rumbaugh, G. SYNGAP1 links the maturation rate of excitatory synapses to the duration of critical-period synaptic plasticity. J. Neurosci. 2013, 33, 10447–10452. [Google Scholar] [CrossRef] [PubMed]
- Ogden, K.K.; Ozkan, E.D.; Rumbaugh, G. Prioritizing the development of mouse models for childhood brain disorders. Neuropharmacology 2016, 100, 2–16. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakajima, R.; Takao, K.; Hattori, S.; Shoji, H.; Komiyama, N.H.; Grant, S.G.N.; Miyakawa, T. Comprehensive behavioral analysis of heterozygous Syngap1 knockout mice. Neuropsychopharmacol. Rep. 2019, 39, 223–237. [Google Scholar] [CrossRef]
- Ozkan, E.D.; Creson, T.K.; Kramár, E.A.; Rojas, C.; Seese, R.R.; Babyan, A.H.; Shi, Y.; Lucero, R.; Xu, X.; Noebels, J.L.; et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron 2014, 82, 1317–1333. [Google Scholar] [CrossRef]
- Zhang, Y.; Pak, C.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Zhang, H.; Powell, S.K.; Fernando, M.B.; Zhang, S.; Flaherty, E.K.; Ho, S.-M.; Slesinger, P.A.; Duan, J.; Brennand, K.J. ASCL1- and DLX2-induced GABAergic neurons from hiPSC-derived NPCs. J. Neurosci. Methods 2020, 334, 108548. [Google Scholar] [CrossRef]
- Blair, J.D.; Bateup, H.S. New frontiers in modeling tuberous sclerosis with human stem cell-derived neurons and brain organoids. Dev. Dyn. 2020, 249, 46–55. [Google Scholar] [CrossRef]
- Afshar Saber, W.; Sahin, M. Recent advances in human stem cell-based modeling of Tuberous Sclerosis Complex. Mol. Autism 2020, 11, 16. [Google Scholar] [CrossRef]
- Costa, V.; Aigner, S.; Vukcevic, M.; Sauter, E.; Behr, K.; Ebeling, M.; Dunkley, T.; Friedlein, A.; Zoffmann, S.; Meyer, C.A.; et al. mTORC1 Inhibition Corrects Neurodevelopmental and Synaptic Alterations in a Human Stem Cell Model of Tuberous Sclerosis. Cell Rep. 2016, 15, 86–95. [Google Scholar] [CrossRef]
- Li, Y.; Cao, J.; Chen, M.; Li, J.; Sun, Y.; Zhang, Y.; Zhu, Y.; Wang, L.; Zhang, C. Abnormal Neural Progenitor Cells Differentiated from Induced Pluripotent Stem Cells Partially Mimicked Development of TSC2 Neurological Abnormalities. Stem Cell Rep. 2017, 8, 883–893. [Google Scholar] [CrossRef]
- Nadadhur, A.G.; Alsaqati, M.; Gasparotto, L.; Cornelissen-Steijger, P.; van Hugte, E.; Dooves, S.; Harwood, A.J.; Heine, V.M. Neuron-Glia Interactions Increase Neuronal Phenotypes in Tuberous Sclerosis Complex Patient iPSC-Derived Models. Stem Cell Rep. 2019, 12, 42–56. [Google Scholar] [CrossRef]
- Zucco, A.J.; Pozzo, V.D.; Afinogenova, A.; Hart, R.P.; Devinsky, O.; D’Arcangelo, G. Neural progenitors derived from Tuberous Sclerosis Complex patients exhibit attenuated PI3K/AKT signaling and delayed neuronal differentiation. Mol. Cell. Neurosci. 2018, 92, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Sundberg, M.; Yang, C.; Wafa, S.M.A.; Dwyer, S.; Chen, P.-F.; Buttermore, E.D.; Sahin, M. Biallelic Mutations in TSC2 Lead to Abnormalities Associated with Cortical Tubers in Human iPSC-Derived Neurons. J. Neurosci. 2019, 39, 9294–9305. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef]
- Grabole, N.; Zhang, J.D.; Aigner, S.; Ruderisch, N.; Costa, V.; Weber, F.C.; Theron, M.; Berntenis, N.; Spleiss, O.; Ebeling, M.; et al. Genomic analysis of the molecular neuropathology of tuberous sclerosis using a human stem cell model. Genome Med. 2016, 8, 94. [Google Scholar] [CrossRef]
- Alsaqati, M.; Heine, V.M.; Harwood, A.J. Pharmacological intervention to restore connectivity deficits of neuronal networks derived from ASD patient iPSC with a TSC2 mutation. Mol. Autism 2020, 11, 80. [Google Scholar] [CrossRef]
- Catlett, T.S.; Onesto, M.M.; McCann, A.J.; Rempel, S.K.; Glass, J.; Franz, D.N.; Gómez, T.M. RHOA signaling defects result in impaired axon guidance in iPSC-derived neurons from patients with tuberous sclerosis complex. Nat. Commun. 2021, 12, 2589. [Google Scholar] [CrossRef] [PubMed]
- Eichmüller, O.L.; Corsini, N.S.; Vértesy, Á.; Scholl, T.; Gruber, V.-E.; Peer, A.M.; Chu, J.; Novatchkova, M.; Paredes, M.F.; Feucht, M.; et al. Cerebral Organoid Model Reveals Excessive Proliferation of Human Caudal Late Interneuron Progenitors in Tuberous Sclerosis Complex. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wong, C.W.; Wang, Y.; Liu, T.; Li, L.; Cheung, S.K.K.; Or, P.M.-Y.; Cheng, A.S.-L.; Choy, K.W.; Burbach, J.P.H.; Feng, B.; et al. Autism-associated PTEN missense mutation leads to enhanced nuclear localization and neurite outgrowth in an induced pluripotent stem cell line. FEBS J. 2020, 287, 4848–4861. [Google Scholar] [CrossRef]
- Spinelli, L.; Black, F.M.; Berg, J.N.; Eickholt, B.J.; Leslie, N.R. Functionally distinct groups of inherited PTEN mutations in autism and tumour syndromes. J. Med. Genet. 2015, 52, 128–134. [Google Scholar] [CrossRef]
- Lee, S.H.; Park, J.-S.; Byun, J.-K.; Jhun, J.; Jung, K.; Seo, H.-B.; Moon, Y.-M.; Kim, H.-Y.; Park, S.-H.; Cho, M.-L. PTEN ameliorates autoimmune arthritis through down-regulating STAT3 activation with reciprocal balance of Th17 and Tregs. Sci. Rep. 2016, 6, 34617. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.A.; Sur, M.; Gehrke, L.; Knoblich, J.A.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef] [PubMed]
- Klofas, L.K.; Short, B.P.; Snow, J.P.; Sinnaeve, J.; Rushing, G.V.; Westlake, G.; Weinstein, W.; Ihrie, R.A.; Ess, K.C.; Carson, R.P. DEPDC5 haploinsufficiency drives increased mTORC1 signaling and abnormal morphology in human iPSC-derived cortical neurons. Neurobiol. Dis. 2020, 143, 104975. [Google Scholar] [CrossRef]
- Mazuelas, H.; Carrió, M.; Serra, E. Modeling tumors of the peripheral nervous system associated with Neurofibromatosis type 1: Reprogramming plexiform neurofibroma cells. Stem Cell Res. 2020, 49, 102068. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Gutmann, D.H. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum. Mol. Genet. 2014, 23, 6712–6721. [Google Scholar] [CrossRef] [PubMed]
- Anastasaki, C.; Woo, A.S.; Messiaen, L.M.; Gutmann, D.H. Elucidating the impact of neurofibromatosis-1 germline mutations on neurofibromin function and dopamine-based learning. Hum. Mol. Genet. 2015, 24, 3518–3528. [Google Scholar] [CrossRef]
- Anastasaki, C.; Wegscheid, M.L.; Hartigan, K.; Papke, J.B.; Kopp, N.D.; Chen, J.; Cobb, O.; Dougherty, J.D.; Gutmann, D.H. Human iPSC-Derived Neurons and Cerebral Organoids Establish Differential Effects of Germline NF1 Gene Mutations. Stem Cell Rep. 2020, 14, 541–550. [Google Scholar] [CrossRef]
- Sagata, N.; Kato, T.A.; Kano, S.-I.; Ohgidani, M.; Shimokawa, N.; Sato-Kasai, M.; Hayakawa, K.; Kuwano, N.; Wilson, A.M.; Ishizuka, K.; et al. Dysregulated gene expressions of MEX3D, FOS and BCL2 in human induced-neuronal (iN) cells from NF1 patients: A pilot study. Sci. Rep. 2017, 7, 13905. [Google Scholar] [CrossRef]
- Lewis, E.M.A.; Meganathan, K.; Baldridge, D.; Gontarz, P.; Zhang, B.; Bonni, A.; Constantino, J.N.; Kroll, K.L. Cellular and molecular characterization of multiplex autism in human induced pluripotent stem cell-derived neurons. Mol. Autism 2019, 10, 51. [Google Scholar] [CrossRef]
- Sagata, N.; Kano, S.-I.; Ohgidani, M.; Inamine, S.; Sakai, Y.; Kato, H.; Masuda, K.; Nakahara, T.; Nakahara-Kido, M.; Ohga, S.; et al. Forskolin rapidly enhances neuron-like morphological change of directly induced-neuronal cells from neurofibromatosis type 1 patients. Neuropsychopharmacol. Rep. 2020, 40, 396–400. [Google Scholar] [CrossRef]
- Sakai, T.; Naito, A.T.; Kuramoto, Y.; Ito, M.; Okada, K.; Higo, T.; Nakagawa, A.; Shibamoto, M.; Yamaguchi, T.; Sumida, T.; et al. Phenotypic Screening Using Patient-Derived Induced Pluripotent Stem Cells Identified Pyr3 as a Candidate Compound for the Treatment of Infantile Hypertrophic Cardiomyopathy. Int. Heart J. 2018, 59, 1096–1105. [Google Scholar] [CrossRef]
- Jaffré, F.; Miller, C.L.; Schänzer, A.; Evans, T.; Roberts, A.E.; Hahn, A.; Kontaridis, M.I. Inducible Pluripotent Stem Cell-Derived Cardiomyocytes Reveal Aberrant Extracellular Regulated Kinase 5 and Mitogen-Activated Protein Kinase Kinase 1/2 Signaling Concomitantly Promote Hypertrophic Cardiomyopathy in RAF1-Associated Noonan Syndrome. Circulation 2019, 140, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Hanses, U.; Kleinsorge, M.; Roos, L.; Yigit, G.; Li, Y.; Barbarics, B.; El-Battrawy, I.; Lan, H.; Tiburcy, M.; Hindmarsh, R.; et al. Intronic CRISPR Repair in a Preclinical Model of Noonan Syndrome-Associated Cardiomyopathy. Circulation 2020, 142, 1059–1076. [Google Scholar] [CrossRef]
- Higgins, E.M.; Bos, J.M.; Dotzler, S.M.; John Kim, C.S.; Ackerman, M.J. MRAS Variants Cause Cardiomyocyte Hypertrophy in Patient-Specific Induced Pluripotent Stem Cell-Derived Cardiomyocytes: Additional Evidence for MRAS as a Definitive Noonan Syndrome-Susceptibility Gene. Circ. Genom. Precis. Med. 2019, 12, e002648. [Google Scholar] [CrossRef]
- Ju, Y.; Park, J.S.; Kim, D.; Kim, B.; Lee, J.H.; Nam, Y.; Yoo, H.-W.; Lee, B.H.; Han, Y.-M. SHP2 mutations induce precocious gliogenesis of Noonan syndrome-derived iPSCs during neural development in vitro. Stem Cell Res. Ther. 2020, 11, 209. [Google Scholar] [CrossRef] [PubMed]
- Han, K.-M.; Kim, S.-K.; Kim, D.; Choi, J.-Y.; Im, I.; Hwang, K.-S.; Kim, C.-H.; Lee, B.H.; Yoo, H.-W.; Han, Y.-M. Enhanced SMAD1 Signaling Contributes to Impairments of Early Development in CFC-iPSCs. Stem Cells 2015, 33, 1447–1455. [Google Scholar] [CrossRef]
- Yeh, E.; Dao, D.Q.; Wu, Z.Y.; Kandalam, S.M.; Camacho, F.M.; Tom, C.; Zhang, W.; Krencik, R.; Rauen, K.A.; Ullian, E.M.; et al. Patient-derived iPSCs show premature neural differentiation and neuron type-specific phenotypes relevant to neurodevelopment. Mol. Psychiatry 2018, 23, 1687–1698. [Google Scholar] [CrossRef] [PubMed]
- Krueger, D.A.; Care, M.M.; Holland, K.; Agricola, K.; Tudor, C.; Mangeshkar, P.; Wilson, K.A.; Byars, A.; Sahmoud, T.; Franz, D.N. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med. 2010, 363, 1801–1811. [Google Scholar] [CrossRef]
- Ostendorf, A.P.; Wong, M. mTOR inhibition in epilepsy: Rationale and clinical perspectives. CNS Drugs 2015, 29, 91–99. [Google Scholar] [CrossRef]
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N.; et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 2153–2163. [Google Scholar] [CrossRef]
- Way, S.W.; Rozas, N.S.; Wu, H.C.; McKenna, J.; Reith, R.M.; Hashmi, S.S.; Dash, P.K.; Gambello, M.J. The differential effects of prenatal and/or postnatal rapamycin on neurodevelopmental defects and cognition in a neuroglial mouse model of tuberous sclerosis complex. Hum. Mol. Genet. 2012, 21, 3226–3236. [Google Scholar] [CrossRef] [PubMed]
- Cox, R.L.; Calderon de Anda, F.; Mangoubi, T.; Yoshii, A. Multiple Critical Periods for Rapamycin Treatment to Correct Structural Defects in Tsc-1-Suppressed Brain. Front. Mol. Neurosci. 2018, 11, 409. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Arriola Apelo, S.I.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.-Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat. Commun. 2019, 10, 3194. [Google Scholar] [CrossRef]
- Rageot, D.; Bohnacker, T.; Keles, E.; McPhail, J.A.; Hoffmann, R.M.; Melone, A.; Borsari, C.; Sriramaratnam, R.; Sele, A.M.; Beaufils, F.; et al. (S)-4-(Difluoromethyl)-5-(4-(3-methylmorpholino)-6-morpholino-1,3,5-triazin-2-yl)pyridin-2-amine (PQR530), a Potent, Orally Bioavailable, and Brain-Penetrable Dual Inhibitor of Class I PI3K and mTOR Kinase. J. Med. Chem. 2019, 62, 6241–6261. [Google Scholar] [CrossRef]
- Borsari, C.; Keles, E.; Treyer, A.; de Pascale, M.; Hebeisen, P.; Hamburger, M.; Wymann, M.P. Second-generation tricyclic pyrimido-pyrrolo-oxazine mTOR inhibitor with predicted blood-brain barrier permeability. RSC Med. Chem. 2021, 12, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Nikolaeva, I.; Kazdoba, T.M.; Crowell, B.; D’Arcangelo, G. Differential roles for Akt and mTORC1 in the hypertrophy of Pten mutant neurons, a cellular model of brain overgrowth disorders. Neuroscience 2017, 354, 196–207. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef]
- Jindal, G.A.; Goyal, Y.; Burdine, R.D.; Rauen, K.A.; Shvartsman, S.Y. RASopathies: Unraveling mechanisms with animal models. Dis. Model. Mech. 2015, 8, 769–782. [Google Scholar] [CrossRef]
- Li, W.; Cui, Y.; Kushner, S.A.; Brown, R.A.M.; Jentsch, J.D.; Frankland, P.W.; Cannon, T.D.; Silva, A.J. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 2005, 15, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Krab, L.C.; de Goede-Bolder, A.; Aarsen, F.K.; Pluijm, S.M.F.; Bouman, M.J.; van der Geest, J.N.; Lequin, M.; Catsman, C.E.; Arts, W.F.M.; Kushner, S.A.; et al. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: A randomized controlled trial. JAMA 2008, 300, 287–294. [Google Scholar] [CrossRef]
- Van der Vaart, T.; Plasschaert, E.; Rietman, A.B.; Renard, M.; Oostenbrink, R.; Vogels, A.; de Wit, M.-C.Y.; Descheemaeker, M.-J.; Vergouwe, Y.; Catsman-Berrevoets, C.E.; et al. Simvastatin for cognitive deficits and behavioural problems in patients with neurofibromatosis type 1 (NF1-SIMCODA): A randomised, placebo-controlled trial. Lancet Neurol. 2013, 12, 1076–1083. [Google Scholar] [CrossRef]
- Payne, J.M.; Barton, B.; Ullrich, N.J.; Cantor, A.; Hearps, S.J.C.; Cutter, G.; Rosser, T.; Walsh, K.S.; Gioia, G.A.; Wolters, P.L.; et al. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 2016, 87, 2575–2584. [Google Scholar] [CrossRef]
- Stivaros, S.; Garg, S.; Tziraki, M.; Cai, Y.; Thomas, O.; Mellor, J.; Morris, A.A.; Jim, C.; Szumanska-Ryt, K.; Parkes, L.M.; et al. Randomised controlled trial of simvastatin treatment for autism in young children with neurofibromatosis type 1 (SANTA). Mol. Autism 2018, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Mainberger, F.; Jung, N.H.; Zenker, M.; Wahlländer, U.; Freudenberg, L.; Langer, S.; Berweck, S.; Winkler, T.; Straube, A.; Heinen, F.; et al. Lovastatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol. 2013, 13, 131. [Google Scholar] [CrossRef] [PubMed]
- Bearden, C.E.; Hellemann, G.S.; Rosser, T.; Montojo, C.; Jonas, R.; Enrique, N.; Pacheco, L.; Hussain, S.A.; Wu, J.Y.; Ho, J.S.; et al. A randomized placebo-controlled lovastatin trial for neurobehavioral function in neurofibromatosis I. Ann. Clin. Transl. Neurol. 2016, 3, 266–279. [Google Scholar] [CrossRef]
- Chen, P.-C.; Wakimoto, H.; Conner, D.; Araki, T.; Yuan, T.; Roberts, A.; Seidman, C.E.; Bronson, R.; Neel, B.G.; Seidman, J.G.; et al. Activation of multiple signaling pathways causes developmental defects in mice with a Noonan syndrome–associated Sos1 mutation. J. Clin. Investig. 2010, 120, 4353–4365. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Simpson, J.; Hong, J.H.; Kim, K.-H.; Thavarajah, N.K.; Backx, P.H.; Neel, B.G.; Araki, T. MEK-ERK pathway modulation ameliorates disease phenotypes in a mouse model of Noonan syndrome associated with the Raf1(L613V) mutation. J. Clin. Investig. 2011, 121, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Wolters, P.L.; Dombi, E.; Baldwin, A.; Whitcomb, P.; Fisher, M.J.; Weiss, B.; Kim, A.; Bornhorst, M.; Shah, A.C.; et al. Selumetinib in Children with Inoperable Plexiform Neurofibromas. N. Engl. J. Med. 2020, 382, 1430–1442. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef]
- Andelfinger, G.; Marquis, C.; Raboisson, M.-J.; Théoret, Y.; Waldmüller, S.; Wiegand, G.; Gelb, B.D.; Zenker, M.; Delrue, M.-A.; Hofbeck, M. Hypertrophic Cardiomyopathy in Noonan Syndrome Treated by MEK-Inhibition. J. Am. Coll. Cardiol. 2019, 73, 2237–2239. [Google Scholar] [CrossRef] [PubMed]
- Pucilowska, J.; Vithayathil, J.; Pagani, M.; Kelly, C.; Karlo, J.C.; Robol, C.; Morella, I.; Gozzi, A.; Brambilla, R.; Landreth, G.E. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J. Neurosci. 2018, 38, 6640–6652. [Google Scholar] [CrossRef] [PubMed]
- Pudewell, S.; Ahmadian, M.R. Spotlight on Accessory Proteins: RTK-RAS-MAPK Modulators as New Therapeutic Targets. Biomolecules 2021, 11, 895. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.G.; Del Río, I.B.; Sari, S.; Sekerim, A.; Young, L.C.; Hartig, N.; Areso Zubiaur, I.; El-Bahrawy, M.A.; Hynds, R.E.; Lei, W.; et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat. Commun. 2019, 10, 2532. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Clinical Phenotypes (Frequency) | Animal Models | Human Cell Models | Mutations in 10,000 ASD Cases [19] | |
|---|---|---|---|---|
| mTOR Pathway | ||||
| TSC1-2 | Tuberous Sclerosis (1:10,000) ASD, ID, epilepsy, structural neurological changes, cancers/tumors, skin problems | Altered synaptic plasticity; altered social behavior; altered learning behavior and memory; disrupted neuronal cell development; seizures | Increased dendritic branching; altered synaptic plasticity; enlarged cell size | TSC1: n.d. TSC2: 3.8 |
| PTEN | Cowden Syndrome (1:250,000) ASD, ID, epilepsy, structural neurological changes, cancers/tumors, skin problems | Altered synaptic plasticity; altered social behavior; altered learning behavior and memory; disrupted neuronal cell development; seizures | Impaired cortical folding | PTEN: 4.4 |
| AKT1-3 | Cowden Syndrome (1–9:1,000,000) and AKT1-related Proteus Syndrome ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Altered synaptic plasticity; altered social behavior; altered learning behavior and memory; disrupted neuronal cell development; seizures | Increased dendritic branching; altered synaptic plasticity; enlarged cell size | AKT1: n.d. AKT2: 1.3 AKT3: 1.3 |
| MTOR | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Disrupted neuronal cell development; seizures | MTOR: 0.6 | |
| RICTOR/RAPTOR | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Altered synaptic plasticity; disrupted neuronal cell development | RICTOR: n.d. RAPTOR: 2.5 | |
| RHEB | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Altered synaptic plasticity; disrupted neuronal cell development; seizures | n.d. | |
| S6K | ASD, ID, epilepsy, structural neurological changes, cancers/tumors, skin problems | Minor altered synaptic plasticity; altered learning behavior and memory; altered social behavior; disrupted neuronal cell development | RPS6KB1: 1.9 | |
| eIF4EeIF4E-B | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Altered synaptic plasticity; altered social behavior; altered learning behavior and memory; disrupted neuronal cell development | EIF4B: 0.6 | |
| DEPDC5 | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | Altered synaptic plasticity; disrupted neuronal cell development; seizures | Enlarged cell size; increased proliferation rates | DEPDC5: 0.6 |
| NPRL2 | ASD, ID, epilepsy, structural neurological changes, cancers/tumors | n.d | ||
| RAS Pathway | ||||
| SYNGAP1 | SYNGAP1-ID ASD, ID, epilepsy, schizophrenia | Altered synaptic plasticity (reduced hippocampal LTP and early spine maturation); elevated protein synthesis; altered spatial learning and memory, social novelty, fear memory and increased locomotor activity | SYNGAP1: 10.8 | |
| NF1 | NF1 Syndrome (1: 3000): ASD, ID, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors, skin problems | Altered synaptic plasticity (reduced hippocampal LTP and spine density); altered spatial learning and memory, social learning, attention and working memory deficits | Altered proliferation, apoptosis and neuronal differentiation | NF1: 4.4 |
| SPRED1 | Legius Syndrome ASD, ID, structural neurological changes., congenital heart disease, skin problems | Altered synaptic plasticity (reduced hippocampal LTP), altered spatial learning and memory and social behavior | SPRED1: 0.6 | |
| SOS1 | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | craniofacial abnormalities; cardiac defects | n.d. | |
| CBL | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | CBL: 0.6 | ||
| HRAS | Costello Syndrome (1:300,000–1.25 mil.) ASD, ID, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors, skin problems | Increased soma size and spine complexity: enhanced LTP; altered spatial learning and memory and contextual fear conditioning | Increased astrogenesis, increased proteoglycans, dysregulated cortical maturation | n.d. |
| NRAS | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Leukemias, craniofacial abnormalities, cardiac defects | n.d. | |
| KRAS | CFC and Noonan Syndrome ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Altered synaptic plasticity (reduced hippocampal LTP); elevated protein synthesis; altered spatial learning and memory, working memory; normal social behaviors | KRAS: 0.6 | |
| RIT1 | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Reduced dendritic length and complexity | n.d. | |
| RAF1 | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Increased astroglial cell density; enhanced spatial learning and memory | n.d. | |
| BRAF | CFC and Noonan Syndrome ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Altered synaptic plasticity (reduced hippocampal LTP); altered spatial learning and memory; seizures | Inhibited neuronal differentiation; premature differentiation and impaired cortical layering | BRAF: 1.3 |
| MEK1/2 | CFC Syndrome (1:800,000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Increased astroglial cells, neuronal loss; altered spatial learning and memory; altered fear conditioning; craniofacial abnormalities | MAP2K1: 0.6MAP2K2: 1.3 | |
| PTPN11 (SHP2) | Noonan Syndrome (1:2000) ASD, ID, epilepsy, structural neurological changes, congenital heart disease, bone malformations, cancers/tumors | Altered synaptic plasticity; altered spatial learning and memory; increased hyperactivity, reduced anxiety behavior | Inhibited differentiation, increased gliogenesis; reduced neurite outgrowth; lower spontaneous firing rate | PTPN11: 1.3 |
| LZTR1 | Noonan Syndrome (1:2000) | LZTR1: 3.2 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vasic, V.; Jones, M.S.O.; Haslinger, D.; Knaus, L.S.; Schmeisser, M.J.; Novarino, G.; Chiocchetti, A.G. Translating the Role of mTOR- and RAS-Associated Signalopathies in Autism Spectrum Disorder: Models, Mechanisms and Treatment. Genes 2021, 12, 1746. https://doi.org/10.3390/genes12111746
Vasic V, Jones MSO, Haslinger D, Knaus LS, Schmeisser MJ, Novarino G, Chiocchetti AG. Translating the Role of mTOR- and RAS-Associated Signalopathies in Autism Spectrum Disorder: Models, Mechanisms and Treatment. Genes. 2021; 12(11):1746. https://doi.org/10.3390/genes12111746
Chicago/Turabian StyleVasic, Verica, Mattson S. O. Jones, Denise Haslinger, Lisa S. Knaus, Michael J. Schmeisser, Gaia Novarino, and Andreas G. Chiocchetti. 2021. "Translating the Role of mTOR- and RAS-Associated Signalopathies in Autism Spectrum Disorder: Models, Mechanisms and Treatment" Genes 12, no. 11: 1746. https://doi.org/10.3390/genes12111746
APA StyleVasic, V., Jones, M. S. O., Haslinger, D., Knaus, L. S., Schmeisser, M. J., Novarino, G., & Chiocchetti, A. G. (2021). Translating the Role of mTOR- and RAS-Associated Signalopathies in Autism Spectrum Disorder: Models, Mechanisms and Treatment. Genes, 12(11), 1746. https://doi.org/10.3390/genes12111746