
Mechanisms of Genome Instability in the Fragile X-Related Disorders

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. FRAXA Chromosome Fragility
3. Repeat Instability
3.1. Repeat Expansions
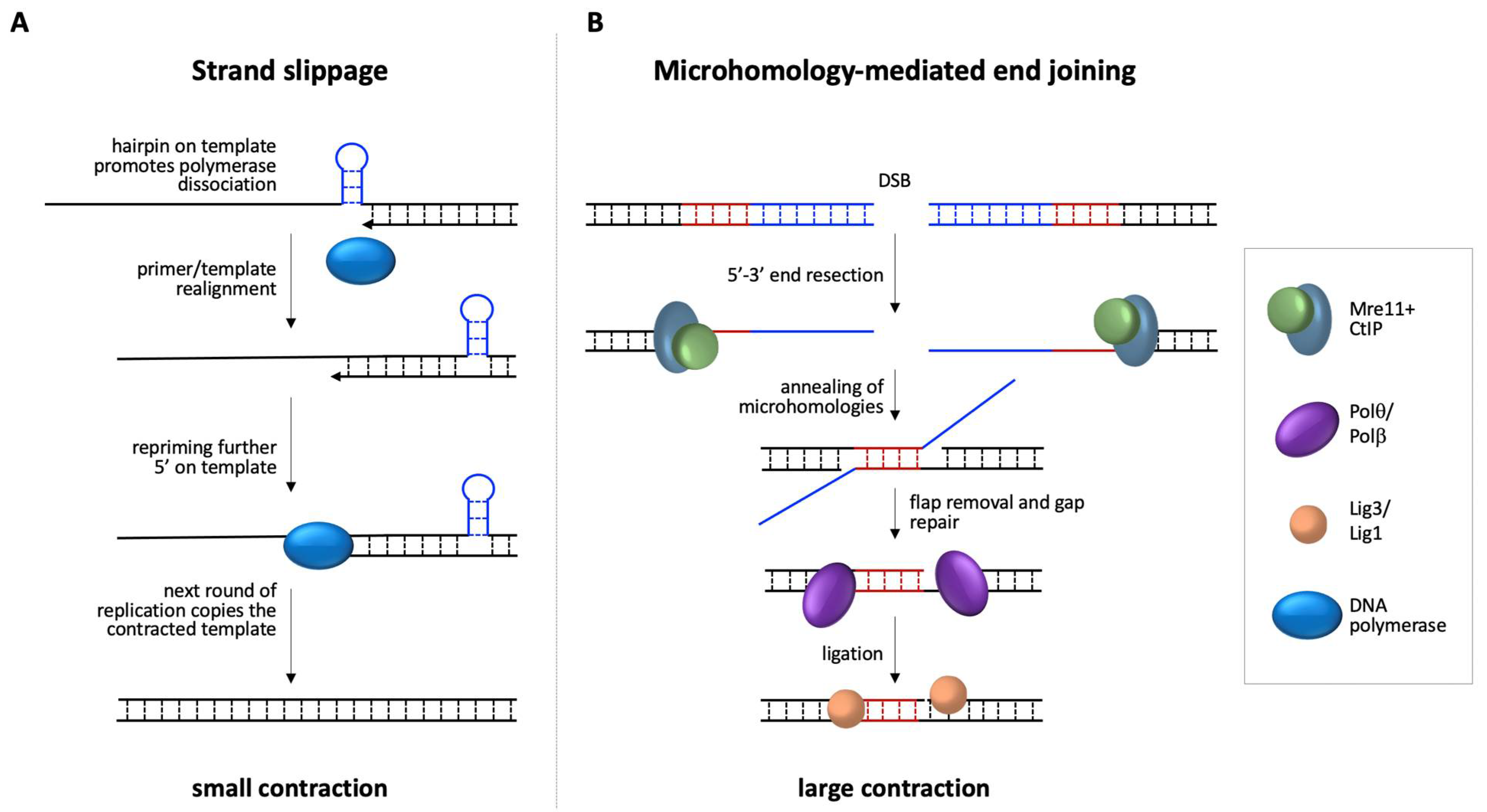
3.2. Repeat Contractions
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Martin, J.P.; Bell, J. A Pedigree of Mental Defect Showing Sex-Linkage. J. Neurol. Psychiatry 1943, 6, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Lubs, H.A. A marker X chromosome. Am. J. Hum. Genet. 1969, 21, 231–244. [Google Scholar]
- Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Pieretti, M.; Sutcliffe, J.S.; Richards, S.; Verkerk, A.J.; Holden, J.J.; Fenwick, R.G., Jr.; Warren, S.T.; et al. Variation of the CGG repeat at the fragile X site results in genetic instability: Resolution of the Sherman paradox. Cell 1991, 67, 1047–1058. [Google Scholar] [CrossRef]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Sherman, S.L.; Morton, N.E.; Jacobs, P.A.; Turner, G. The marker (X) syndrome: A cytogenetic and genetic analysis. Ann. Hum. Genet. 1984, 48, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Rosero, C.A.; Hagerman, R.J. Fragile X spectrum disorders. Intractable Rare Dis. Res. 2014, 3, 134–146. [Google Scholar] [CrossRef]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Grasso, M.; Faravelli, F.; Lo Nigro, C.; Chiurazzi, P.; Sperandeo, M.P.; Argusti, A.; Pomponi, M.G.; Lecora, M.; Sebastio, G.F.; Perroni, L.; et al. Mosaicism for the full mutation and a microdeletion involving the CGG repeat and flanking sequences in the FMR1 gene in eight fragile X patients. Am. J. Med. Genet. 1999, 85, 311–316. [Google Scholar] [CrossRef]
- Manor, E.; Jabareen, A.; Magal, N.; Kofman, A.; Hagerman, R.J.; Tassone, F. Prenatal Diagnosis of Fragile X: Can a Full Mutation Allele in the FMR1 Gene Contract to a Normal Size? Front. Genet. 2017, 8, 158. [Google Scholar] [CrossRef]
- Prawer, Y.; Hunter, M.; Cronin, S.; Ling, L.; Aliaga Vera, S.; Fahey, M.; Gelfand, N.; Oertel, R.; Bartlett, E.; Francis, D.; et al. Prenatal Diagnosis of Fragile X Syndrome in a Twin Pregnancy Complicated by a Complete Retraction. Genes 2018, 9, 287. [Google Scholar] [CrossRef]
- Maia, N.; Loureiro, J.R.; Oliveira, B.; Marques, I.; Santos, R.; Jorge, P.; Martins, S. Contraction of fully expanded FMR1 alleles to the normal range: Predisposing haplotype or rare events? J. Hum. Genet. 2017, 62, 269–275. [Google Scholar] [CrossRef]
- Grønskov, K.; Hjalgrim, H.; Bjerager, M.O.; Brondum-Nielsen, K. Deletion of all CGG repeats plus flanking sequences in FMR1 does not abolish gene expression. Am. J. Hum. Genet. 1997, 61, 961–967. [Google Scholar] [CrossRef]
- de Graaff, E.; de Vries, B.B.; Willemsen, R.; van Hemel, J.O.; Mohkamsing, S.; Oostra, B.A.; van den Ouweland, A.M. The fragile X phenotype in a mosaic male with a deletion showing expression of the FMR1 protein in 28% of the cells. Am. J. Med. Genet. 1996, 64, 302–308. [Google Scholar] [CrossRef]
- Erbs, E.; Fenger-Gron, J.; Jacobsen, C.M.; Lildballe, D.L.; Rasmussen, M. Spontaneous rescue of a FMR1 repeat expansion and review of deletions in the FMR1 non-coding region. Eur. J. Med. Genet. 2021, 64, 104244. [Google Scholar] [CrossRef]
- Tabolacci, E.; Pietrobono, R.; Maneri, G.; Remondini, L.; Nobile, V.; Della Monica, M.; Pomponi, M.G.; Genuardi, M.; Neri, G.; Chiurazzi, P. Reversion to Normal of FMR1 Expanded Alleles: A Rare Event in Two Independent Fragile X Syndrome Families. Genes 2020, 11, 248. [Google Scholar] [CrossRef]
- Hirst, M.; Grewal, P.; Flannery, A.; Slatter, R.; Maher, E.; Barton, D.; Fryns, J.P.; Davies, K. Two new cases of FMR1 deletion associated with mental impairment. Am. J. Hum. Genet. 1995, 56, 67–74. [Google Scholar] [PubMed]
- MacKenzie, J.J.; Sumargo, I.; Taylor, S.A. A cryptic full mutation in a male with a classical fragile X phenotype. Clin. Genet. 2006, 70, 39–42. [Google Scholar] [CrossRef]
- Orrico, A.; Galli, L.; Dotti, M.T.; Plewnia, K.; Censini, S.; Federico, A. Mosaicism for full mutation and normal-sized allele of the FMR1 gene: A new case. Am. J. Med. Genet. 1998, 78, 341–344. [Google Scholar] [CrossRef]
- Ferreira, S.I.; Pires, L.M.; Ferrao, J.; Sa, J.; Serra, A.; Carreira, I.M. Mosaicism for FMR1 gene full mutation and intermediate allele in a female foetus: A postzygotic retraction event. Gene 2013, 527, 421–425. [Google Scholar] [CrossRef]
- Hayward, B.; Loutaev, I.; Ding, X.; Nolin, S.L.; Thurm, A.; Usdin, K.; Smith, C.B. Fragile X syndrome in a male with methylated premutation alleles and no detectable methylated full mutation alleles. Am. J. Med. Genet. A 2019, 179, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Latham, G.J.; Coppinger, J.; Hadd, A.G.; Nolin, S.L. The role of AGG interruptions in fragile X repeat expansions: A twenty-year perspective. Front. Genet. 2014, 5, 244. [Google Scholar] [CrossRef]
- Cohen, I.L.; Nolin, S.L.; Sudhalter, V.; Ding, X.H.; Dobkin, C.S.; Brown, W.T. Mosaicism for the FMR1 gene influences adaptive skills development in fragile X-affected males. Am. J. Med. Genet. 1996, 64, 365–369. [Google Scholar] [CrossRef]
- Han, X.D.; Powell, B.R.; Phalin, J.L.; Chehab, F.F. Mosaicism for a full mutation, premutation, and deletion of the CGG repeats results in 22% FMRP and elevated FMR1 mRNA levels in a high-functioning fragile X male. Am. J. Med. Genet. A 2006, 140, 1463–1471. [Google Scholar] [CrossRef] [PubMed]
- Pretto, D.; Yrigollen, C.M.; Tang, H.T.; Williamson, J.; Espinal, G.; Iwahashi, C.K.; Durbin-Johnson, B.; Hagerman, R.J.; Hagerman, P.J.; Tassone, F. Clinical and molecular implications of mosaicism in FMR1 full mutations. Front. Genet. 2014, 5, 318. [Google Scholar] [CrossRef] [PubMed]
- Verdyck, P.; Berckmoes, V.; De Vos, A.; Verpoest, W.; Liebaers, I.; Bonduelle, M.; De Rycke, M. Chromosome fragility at FRAXA in human cleavage stage embryos at risk for fragile X syndrome. Am. J. Med. Genet. A 2015, 167, 2306–2313. [Google Scholar] [CrossRef]
- Dobkin, C.; Radu, G.; Ding, X.H.; Brown, W.T.; Nolin, S.L. Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: Fragile X leads to chromosome loss. Am. J. Med. Genet. A 2009, 149, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, R.A.; Kumari, D.; Usdin, K. Common Threads: Aphidicolin-Inducible and Folate-Sensitive Fragile Sites in the Human Genome. Front. Genet. 2021, 12, 1508. [Google Scholar] [CrossRef] [PubMed]
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123. [Google Scholar] [CrossRef]
- Khristich, A.N.; Mirkin, S.M. On the wrong DNA track: Molecular mechanisms of repeat-mediated genome instability. J. Biol. Chem. 2020, 295, 4134–4170. [Google Scholar] [CrossRef]
- Brown, R.E.; Freudenreich, C.H. Structure-forming repeats and their impact on genome stability. Curr. Opin. Genet. Dev. 2021, 67, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Polleys, E.J.; Freudenreich, C.H. Homologous recombination within repetitive DNA. Curr. Opin. Genet. Dev. 2021, 71, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, D.; Hayward, B.E.; Aladjem, M.I.; Kumari, D.; Usdin, K. Chromosome fragility and the abnormal replication of the FMR1 locus in fragile X syndrome. Hum. Mol. Genet. 2014, 23, 2940–2952. [Google Scholar] [CrossRef] [PubMed]
- Hansen, R.S.; Canfield, T.K.; Lamb, M.M.; Gartler, S.M.; Laird, C.D. Association of fragile X syndrome with delayed replication of the FMR1 gene. Cell 1993, 73, 1403–1409. [Google Scholar] [CrossRef]
- Webb, T. Delayed replication of Xq27 in individuals with the fragile X syndrome. Am. J. Med. Genet. 1992, 43, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Miller, B.J.; Cross, D.R.; McGarrity, L.J.; Morris, S.M. The essentiality of folate for the maintenance of deoxynucleotide precursor pools, DNA synthesis, and cell cycle progression in PHA-stimulated lymphocytes. Environ. Health Perspect 1993, 101 (Suppl. 5), 173–178. [Google Scholar] [CrossRef]
- Usdin, K.; Woodford, K.J. CGG repeats associated with DNA instability and chromosome fragility form structures that block DNA synthesis in vitro. Nucleic Acids Res. 1995, 23, 4202–4209. [Google Scholar] [CrossRef]
- Fry, M.; Loeb, L.A. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc. Natl. Acad. Sci. USA 1994, 91, 4950–4954. [Google Scholar] [CrossRef]
- Mitas, M.; Yu, A.; Dill, J.; Haworth, I.S. The trinucleotide repeat sequence d(CGG)15 forms a heat-stable hairpin containing Gsyn. Ganti base pairs. Biochemistry 1995, 34, 12803–12811. [Google Scholar] [CrossRef]
- Yu, A.; Barron, M.D.; Romero, R.M.; Christy, M.; Gold, B.; Dai, J.; Gray, D.M.; Haworth, I.S.; Mitas, M. At physiological pH, d(CCG)15 forms a hairpin containing protonated cytosines and a distorted helix. Biochemistry 1997, 36, 3687–3699. [Google Scholar] [CrossRef]
- Kettani, A.; Kumar, R.A.; Patel, D.J. Solution structure of a DNA quadruplex containing the fragile X syndrome triplet repeat. J. Mol. Biol. 1995, 254, 638–656. [Google Scholar] [CrossRef]
- Murat, P.; Guilbaud, G.; Sale, J.E. DNA polymerase stalling at structured DNA constrains the expansion of short tandem repeats. Genome Biol. 2020, 21, 209. [Google Scholar] [CrossRef]
- Patel, P.K.; Bhavesh, N.S.; Hosur, R.V. Cation-dependent conformational switches in d-TGGCGGC containing two triplet repeats of Fragile X Syndrome: NMR observations. Biochem. Biophys. Res. Commun. 2000, 278, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Fojtik, P.; Vorlickova, M. The fragile X chromosome (GCC) repeat folds into a DNA tetraplex at neutral pH. Nucleic Acids Res. 2001, 29, 4684–4690. [Google Scholar] [CrossRef]
- Mariappan, S.V.; Catasti, P.; Chen, X.; Ratliff, R.; Moyzis, R.K.; Bradbury, E.M.; Gupta, G. Solution structures of the individual single strands of the fragile X DNA triplets (GCC)n.(GGC)n. Nucleic Acids Res. 1996, 24, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Voineagu, I.; Surka, C.F.; Shishkin, A.A.; Krasilnikova, M.M.; Mirkin, S.M. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat. Struct. Mol. Biol. 2009, 16, 226–228. [Google Scholar] [CrossRef]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.R.; Schildkraut, C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 2014, 53, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Bjerregaard, V.A.; Garribba, L.; McMurray, C.T.; Hickson, I.D.; Liu, Y. Folate deficiency drives mitotic missegregation of the human FRAXA locus. Proc. Natl. Acad. Sci. USA 2018, 115, 13003–13008. [Google Scholar] [CrossRef] [PubMed]
- Garribba, L.; Bjerregaard, V.A.; Goncalves Dinis, M.M.; Ozer, O.; Wu, W.; Sakellariou, D.; Pena-Diaz, J.; Hickson, I.D.; Liu, Y. Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl. Acad. Sci. USA 2020, 117, 16527–16536. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Tortora, N.; Allen, E.; Macpherson, J.; Mila, M.; Vianna-Morgante, A.M.; Sherman, S.L.; Dobkin, C.; Latham, G.J.; et al. Expansions and contractions of the FMR1 CGG repeat in 5,508 transmissions of normal, intermediate, and premutation alleles. Am. J. Med. Genet. A 2019, 179, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Nolin, S.L.; Houck, G.E., Jr.; Gargano, A.D.; Blumstein, H.; Dobkin, C.S.; Brown, W.T. FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am. J. Hum. Genet. 1999, 65, 680–688. [Google Scholar] [CrossRef]
- Glaser, D.; Wohrle, D.; Salat, U.; Vogel, W.; Steinbach, P. Mitotic behavior of expanded CGG repeats studied on cultured cells: Further evidence for methylation-mediated triplet repeat stability in fragile X syndrome. Am. J. Med. Genet. 1999, 84, 226–228. [Google Scholar] [CrossRef]
- Wohrle, D.; Salat, U.; Hameister, H.; Vogel, W.; Steinbach, P. Demethylation, reactivation, and destabilization of human fragile X full-mutation alleles in mouse embryocarcinoma cells. Am. J. Hum. Genet. 2001, 69, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gazy, I.; Hayward, B.; Pintado, E.; Hwang, Y.H.; Tassone, F.; Usdin, K. Repeat Instability in the Fragile X-Related Disorders: Lessons from a Mouse Model. Brain Sci. 2019, 9, 52. [Google Scholar] [CrossRef]
- Gazy, I.; Hayward, B.; Potapova, S.; Zhao, X.; Usdin, K. Double-strand break repair plays a role in repeat instability in a fragile X mouse model. DNA Repair 2019, 74, 63–69. [Google Scholar] [CrossRef]
- Møllersen, L.; Rowe, A.D.; Larsen, E.; Rognes, T.; Klungland, A. Continuous and periodic expansion of CAG repeats in Huntington’s disease R6/1 mice. PLoS Genet. 2010, 6, e1001242. [Google Scholar] [CrossRef]
- Zhou, Y.; Kumari, D.; Sciascia, N.; Usdin, K. CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol. Autism. 2016, 7, 42. [Google Scholar] [CrossRef]
- Kennedy, L.; Shelbourne, P.F. Dramatic mutation instability in HD mouse striatum: Does polyglutamine load contribute to cell-specific vulnerability in Huntington’s disease? Hum. Mol. Genet. 2000, 9, 2539–2544. [Google Scholar] [CrossRef]
- Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell 2019, 178, 887–900. [Google Scholar] [CrossRef]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018, 11, dmm031930. [Google Scholar] [CrossRef]
- Kratz, K.; Artola-Boran, M.; Kobayashi-Era, S.; Koh, G.; Oliveira, G.; Kobayashi, S.; Oliveira, A.; Zou, X.; Richter, J.; Tsuda, M.; et al. FANCD2-Associated Nuclease 1 Partially Compensates for the Lack of Exonuclease 1 in Mismatch Repair. Mol. Cell Biol. 2021, 41, e0030321. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. FAN1 protects against repeat expansions in a Fragile X mouse model. DNA Repair 2018, 69, 1–5. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Wilkins, K.; Edelmann, W.; Usdin, K. MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet. 2018, 14, e1007719. [Google Scholar] [CrossRef]
- Zhao, X.N.; Lokanga, R.; Allette, K.; Gazy, I.; Wu, D.; Usdin, K. A MutSbeta-Dependent Contribution of MutSalpha to Repeat Expansions in Fragile X Premutation Mice? PLoS Genet. 2016, 12, e1006190. [Google Scholar] [CrossRef]
- Zhao, X.N.; Kumari, D.; Gupta, S.; Wu, D.; Evanitsky, M.; Yang, W.; Usdin, K. Mutsbeta generates both expansions and contractions in a mouse model of the Fragile X-associated disorders. Hum. Mol. Genet. 2015, 24, 7087–7096. [Google Scholar] [CrossRef] [PubMed]
- Lokanga, A.R.; Zhao, X.N.; Entezam, A.; Usdin, K. X inactivation plays a major role in the gender bias in somatic expansion in a mouse model of the fragile X-related disorders: Implications for the mechanism of repeat expansion. Hum. Mol. Genet. 2014, 23, 4985–4994. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.N.; Usdin, K. Timing of Expansion of Fragile X Premutation Alleles During Intergenerational Transmission in a Mouse Model of the Fragile X-Related Disorders. Front. Genet. 2018, 9, 314. [Google Scholar] [CrossRef]
- Lokanga, R.A.; Zhao, X.N.; Usdin, K. The mismatch repair protein MSH2 is rate limiting for repeat expansion in a fragile X premutation mouse model. Hum. Mutat. 2014, 35, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hayward, B.E.; Steinbach, P.J.; Usdin, K. A point mutation in the nuclease domain of MLH3 eliminates repeat expansions in a mouse stem cell model of the Fragile X-related disorders. Nucleic Acids Res. 2020, 48, 7856–7863. [Google Scholar] [CrossRef]
- Lipkin, S.M.; Moens, P.B.; Wang, V.; Lenzi, M.; Shanmugarajah, D.; Gilgeous, A.; Thomas, J.; Cheng, J.; Touchman, J.W.; Green, E.D.; et al. Meiotic arrest and aneuploidy in MLH3-deficient mice. Nat. Genet. 2002, 31, 385–390. [Google Scholar] [CrossRef]
- Cannavo, E.; Sanchez, A.; Anand, R.; Ranjha, L.; Hugener, J.; Adam, C.; Acharya, A.; Weyland, N.; Aran-Guiu, X.; Charbonnier, J.B.; et al. Regulation of the MLH1-MLH3 endonuclease in meiosis. Nature 2020, 586, 618–622. [Google Scholar] [CrossRef]
- Miller, C.J.; Kim, G.Y.; Zhao, X.; Usdin, K. All three mammalian MutL complexes are required for repeat expansion in a mouse cell model of the Fragile X-related disorders. PLoS Genet. 2020, 16, e1008902. [Google Scholar] [CrossRef]
- Sweder, K.S.; Hanawalt, P.C. Transcription-coupled DNA repair. Science 1993, 262, 439–440. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. Gender and cell-type-specific effects of the transcription-coupled repair protein, ERCC6/CSB, on repeat expansion in a mouse model of the fragile X-related disorders. Hum. Mutat. 2014, 35, 341–349. [Google Scholar] [CrossRef]
- Zhao, X.N.; Usdin, K. The transcription-coupled repair protein ERCC6/CSB also protects against repeat expansion in a mouse model of the fragile X premutation. Hum. Mutat. 2015, 36, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Lufino, M.M.; Wade-Martins, R.; Gromak, N. R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet. 2014, 10, e1004318. [Google Scholar] [CrossRef] [PubMed]
- Loomis, E.W.; Sanz, L.A.; Chedin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Kumari, D.; Usdin, K. Sustained expression of FMR1 mRNA from reactivated fragile X syndrome alleles after treatment with small molecules that prevent trimethylation of H3K27. Hum. Mol. Genet. 2016, 25, 3689–3698. [Google Scholar] [CrossRef]
- Abu Diab, M.; Mor-Shaked, H.; Cohen, E.; Cohen-Hadad, Y.; Ram, O.; Epsztejn-Litman, S.; Eiges, R. The G-rich Repeats in FMR1 and C9orf72 Loci Are Hotspots for Local Unpairing of DNA. Genetics 2018, 210, 1239–1252. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Teng, Y.; Yadav, T.; Duan, M.; Tan, J.; Xiang, Y.; Gao, B.; Xu, J.; Liang, Z.; Liu, Y.; Nakajima, S.; et al. ROS-induced R loops trigger a transcription-coupled but BRCA1/2-independent homologous recombination pathway through CSB. Nat. Commun. 2018, 9, 4115. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Møllersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef]
- Kadyrova, L.Y.; Gujar, V.; Burdett, V.; Modrich, P.L.; Kadyrov, F.A. Human MutLgamma, the MLH1-MLH3 heterodimer, is an endonuclease that promotes DNA expansion. Proc. Natl. Acad. Sci. USA 2020, 117, 3535–3542. [Google Scholar] [CrossRef] [PubMed]
- Kononenko, A.V.; Ebersole, T.; Vasquez, K.M.; Mirkin, S.M. Mechanisms of genetic instability caused by (CGG)n repeats in an experimental mammalian system. Nat. Struct. Mol. Biol. 2018, 25, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Guenolé, A.; Marnef, A.; Clouaire, T.; Puget, N.; Rocher, V.; Arnould, C.; Aguirrebengoa, M.; Genais, M.; Vernekar, D.; et al. BLM-dependent Break-Induced Replication handles Double-strand breaks in transcribed chromatin upon impaired RNA:DNA hybrids dissolution. bioRxiv 2005. [Google Scholar] [CrossRef]
- Marnef, A.; Legube, G. R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol. 2021, 23, 305–313. [Google Scholar] [CrossRef]
- Nakamori, M.; Panigrahi, G.B.; Lanni, S.; Gall-Duncan, T.; Hayakawa, H.; Tanaka, H.; Luo, J.; Otabe, T.; Li, J.; Sakata, A.; et al. A slipped-CAG DNA-binding small molecule induces trinucleotide-repeat contractions in vivo. Nat. Genet. 2020, 52, 146–159. [Google Scholar] [CrossRef]
- Su, X.A.; Freudenreich, C.H. Cytosine deamination and base excision repair cause R-loop-induced CAG repeat fragility and instability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2017, 114, E8392–E8401. [Google Scholar] [CrossRef]
- Kim, J.C.; Harris, S.T.; Dinter, T.; Shah, K.A.; Mirkin, S.M. The role of break-induced replication in large-scale expansions of (CAG)n/(CTG)n repeats. Nat. Struct. Mol. Biol. 2017, 24, 55–60. [Google Scholar] [CrossRef]
- Goldmann, J.M.; Veltman, J.A.; Gilissen, C. De Novo Mutations Reflect Development and Aging of the Human Germline. Trends Genet. 2019, 35, 828–839. [Google Scholar] [CrossRef]
- Reyniers, E.; Vits, L.; De Boulle, K.; Van Roy, B.; Van Velzen, D.; de Graaff, E.; Verkerk, A.J.; Jorens, H.Z.; Darby, J.K.; Oostra, B.; et al. The full mutation in the FMR-1 gene of male fragile X patients is absent in their sperm. Nat. Genet. 1993, 4, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Quan, F.; Grompe, M.; Jakobs, P.; Popovich, B.W. Spontaneous deletion in the FMR1 gene in a patient with fragile X syndrome and cherubism. Hum. Mol. Genet. 1995, 4, 1681–1684. [Google Scholar] [CrossRef] [PubMed]
- de Graaff, E.; Rouillard, P.; Willems, P.J.; Smits, A.P.; Rousseau, F.; Oostra, B.A. Hotspot for deletions in the CGG repeat region of FMR1 in fragile X patients. Hum. Mol. Genet. 1995, 4, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, B.; Ballhausen, W.G.; Pfeiffer, R.A. Mosaicism of a microdeletion of 486 bp involving the CGG repeat of the FMR1 gene due to misalignment of GTT tandem repeats at chi-like elements flanking both breakpoints and a full mutation. Hum. Genet. 1996, 98, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Mila, M.; Castellvi-Bel, S.; Sanchez, A.; Lazaro, C.; Villa, M.; Estivill, X. Mosaicism for the fragile X syndrome full mutation and deletions within the CGG repeat of the FMR1 gene. J. Med. Genet. 1996, 33, 338–340. [Google Scholar] [CrossRef] [PubMed]
- Mannermaa, A.; Pulkkinen, L.; Kajanoja, E.; Ryynanen, M.; Saarikoski, S. Deletion in the FMR1 gene in a fragile-X male. Am. J. Med. Genet. 1996, 64, 293–295. [Google Scholar] [CrossRef]
- Fan, H.; Booker, J.K.; McCandless, S.E.; Shashi, V.; Fleming, A.; Farber, R.A. Mosaicism for an FMR1 gene deletion in a fragile X female. Am. J. Med. Genet. A 2005, 136, 214–217. [Google Scholar] [CrossRef]
- Goncalves, T.F.; dos Santos, J.M.; Goncalves, A.P.; Tassone, F.; Mendoza-Morales, G.; Ribeiro, M.G.; Kahn, E.; Boy, R.; Pimentel, M.M.; Santos-Reboucas, C.B. Finding FMR1 mosaicism in Fragile X syndrome. Expert Rev. Mol. Diagn. 2016, 16, 501–507. [Google Scholar] [CrossRef]
- Luo, S.; Huang, W.; Chen, C.; Pan, Q.; Duan, R.; Wu, L. An novel deletion to normal size in the sperm of a fragile X full mutation male. Clin. Genet. 2014, 86, 295–297. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef]
- Cecconi, M.; Forzano, F.; Rinaldi, R.; Cappellacci, S.; Grammatico, P.; Faravelli, F.; Dagna Bricarelli, F.; Di Maria, E.; Grasso, M. A single nucleotide variant in the FMR1 CGG repeat results in a “Pseudodeletion” and is not associated with the fragile X syndrome phenotype. J. Mol. Diagn. 2008, 10, 272–275. [Google Scholar] [CrossRef]
- Tabolacci, E.; Pomponi, M.G.; Pietrobono, R.; Chiurazzi, P.; Neri, G. A unique case of reversion to normal size of a maternal premutation FMR1 allele in a normal boy. Eur. J. Hum. Genet. 2008, 16, 209–214. [Google Scholar] [CrossRef]
- Tarleton, J.; Kenneson, A.; Taylor, A.K.; Crandall, K.; Fletcher, R.; Casey, R.; Hart, P.S.; Hatton, D.; Fisch, G.; Warren, S.T. A single base alteration in the CGG repeat region of FMR1: Possible effects on gene expression and phenotype. J. Med. Genet. 2002, 39, 196–200. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suelves, N.; Kirkham-McCarthy, L.; Lahue, R.S.; Gines, S. A selective inhibitor of histone deacetylase 3 prevents cognitive deficits and suppresses striatal CAG repeat expansions in Huntington’s disease mice. Sci. Rep. 2017, 7, 6082. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayward, B.E.; Usdin, K. Mechanisms of Genome Instability in the Fragile X-Related Disorders. Genes 2021, 12, 1633. https://doi.org/10.3390/genes12101633
Hayward BE, Usdin K. Mechanisms of Genome Instability in the Fragile X-Related Disorders. Genes. 2021; 12(10):1633. https://doi.org/10.3390/genes12101633
Chicago/Turabian StyleHayward, Bruce E., and Karen Usdin. 2021. "Mechanisms of Genome Instability in the Fragile X-Related Disorders" Genes 12, no. 10: 1633. https://doi.org/10.3390/genes12101633
APA StyleHayward, B. E., & Usdin, K. (2021). Mechanisms of Genome Instability in the Fragile X-Related Disorders. Genes, 12(10), 1633. https://doi.org/10.3390/genes12101633