
Identification of Novel Endogenous Controls for qPCR Normalization in SK-BR-3 Breast Cancer Cell Line



Abstract
1. Introduction
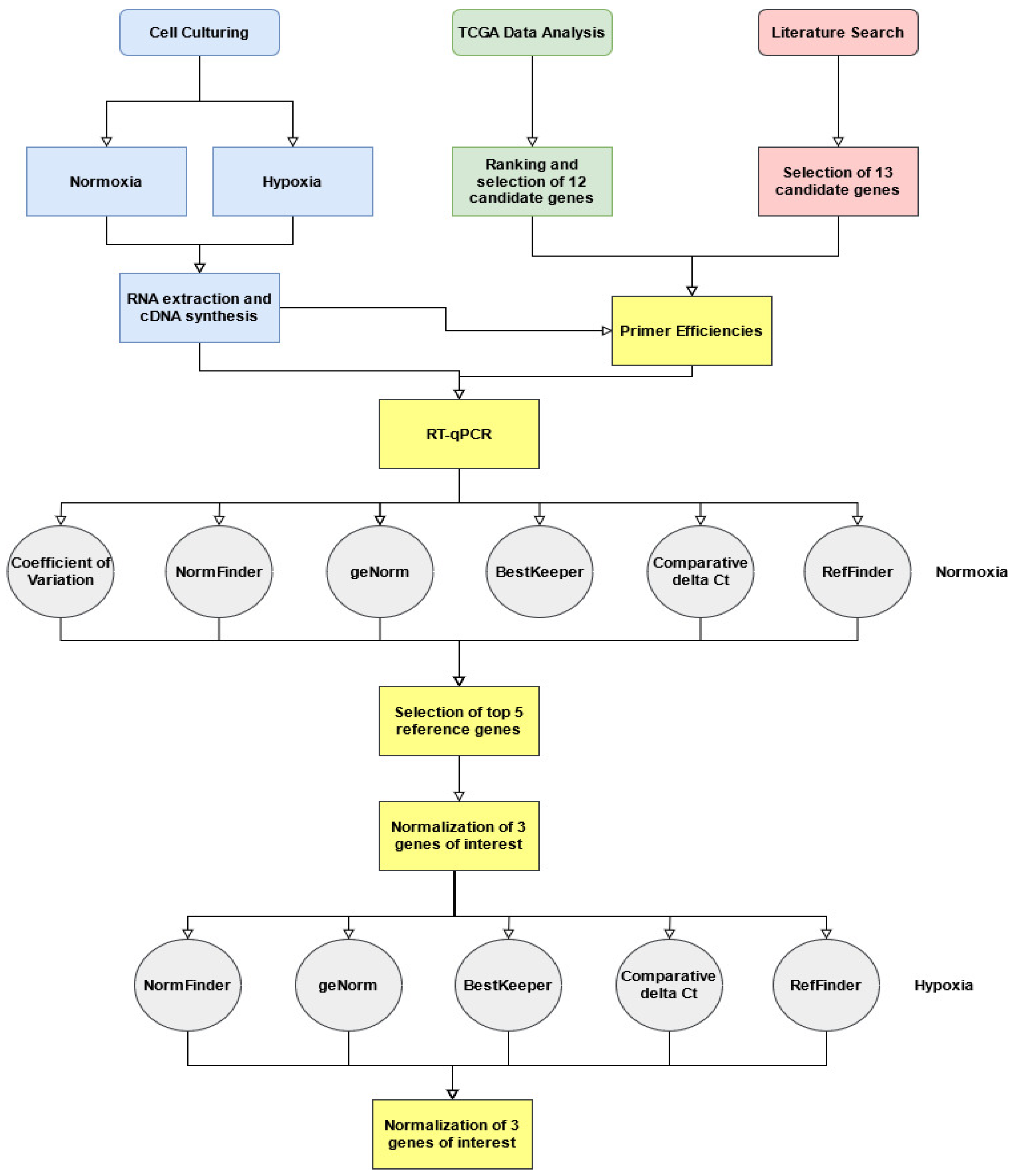
2. Materials and Methods
2.1. TCGA Transcriptomic Analysis for Selection of Novel Candidate Reference Genes
2.2. Gene Ontology (GO)
2.3. Culture and Seeding Conditions
2.4. RNA Extraction and cDNA Synthesis
2.5. Selection of Reference Genes and Primer Design
2.6. Primer Efficiency
2.7. Reverse Transcription Quantitative PCR (RT-qPCR)
2.8. Determination of the Least Variable Reference Genes and Validation in Hypoxic Conditions
3. Results
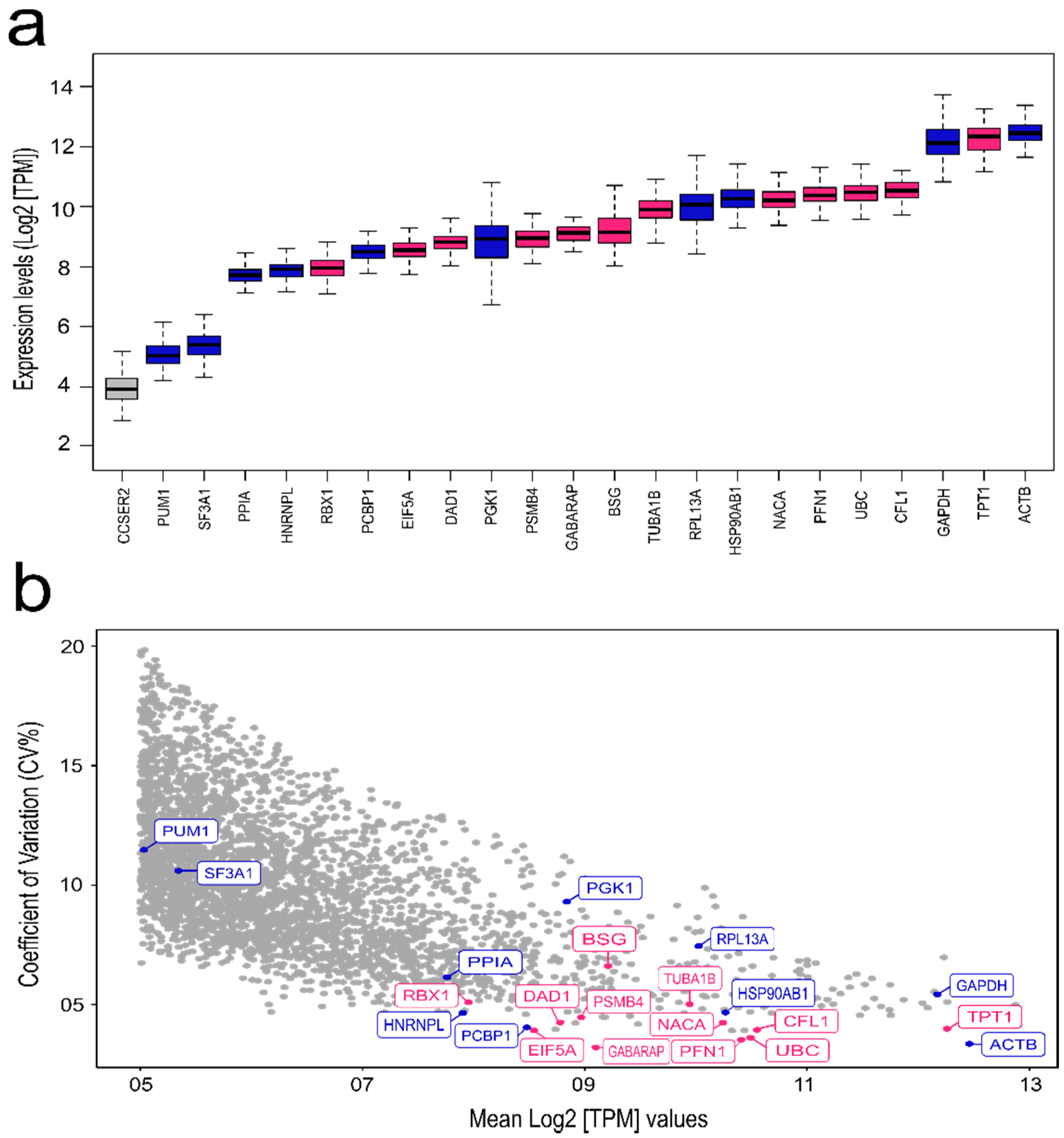
3.1. TCGA Analysis for Selection of Novel Reference Genes
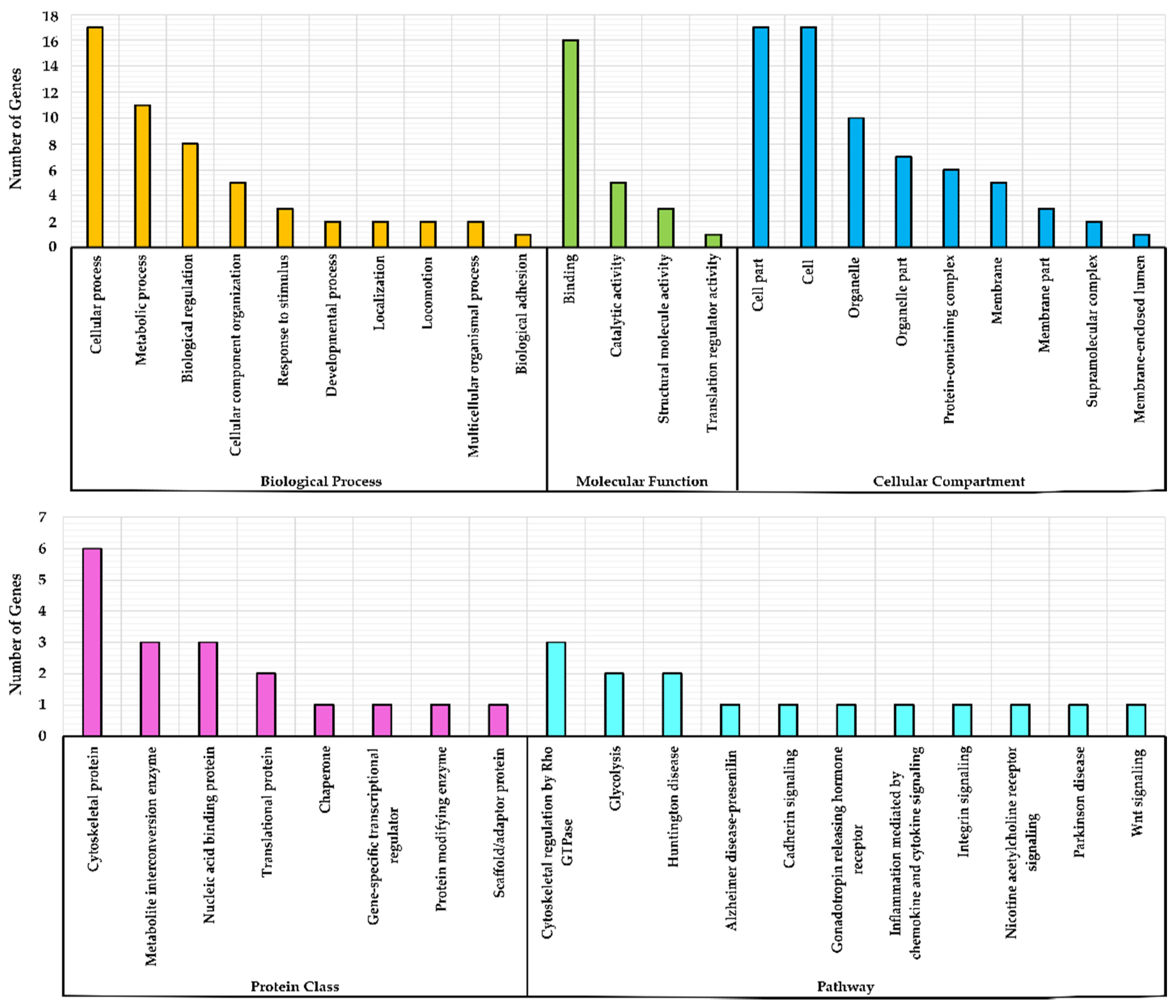
3.2. Gene Ontology (GO) Over-Representation Analysis
3.3. Grouping of Genes Based on Functional Classification (Panther)
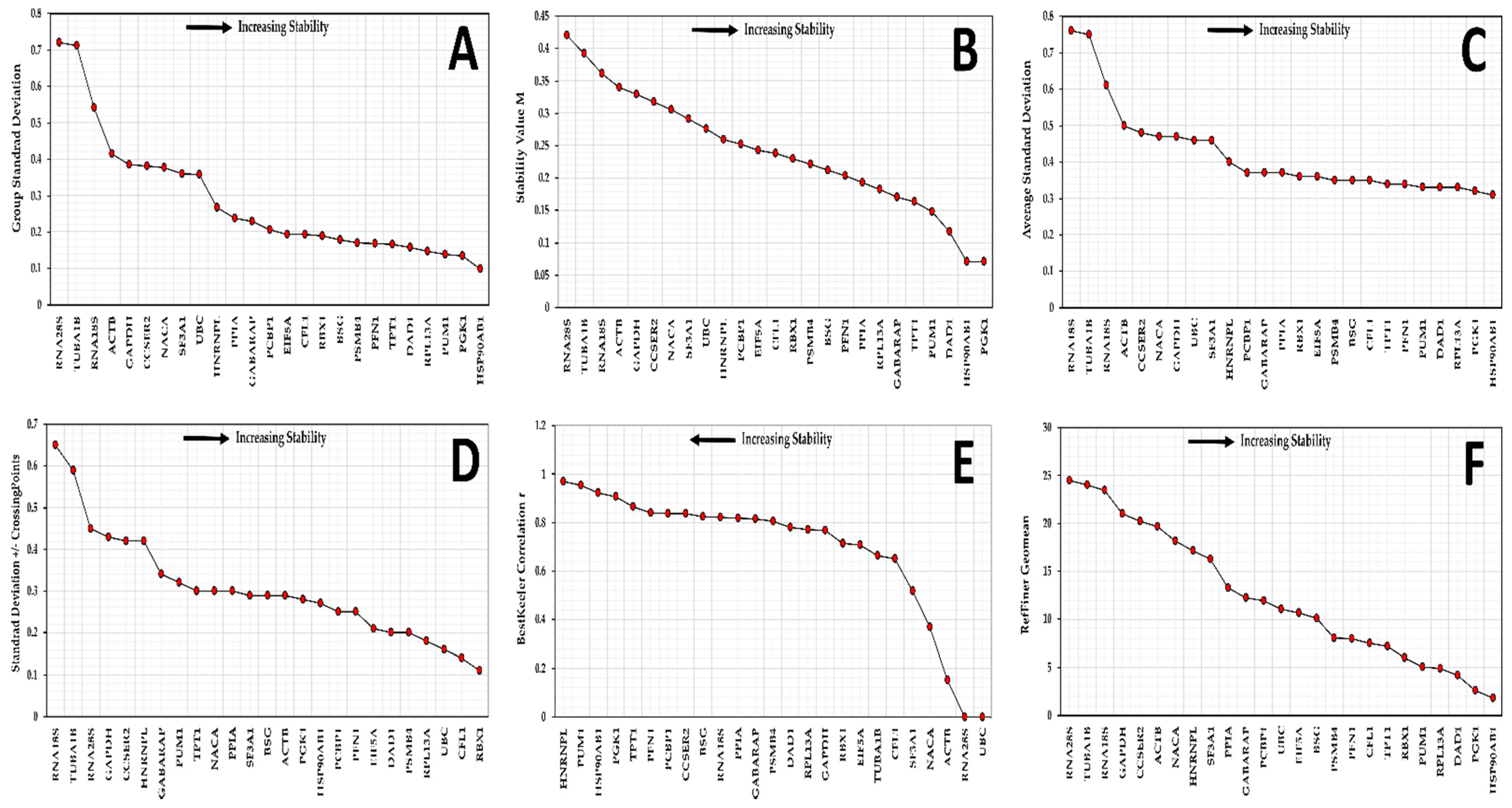
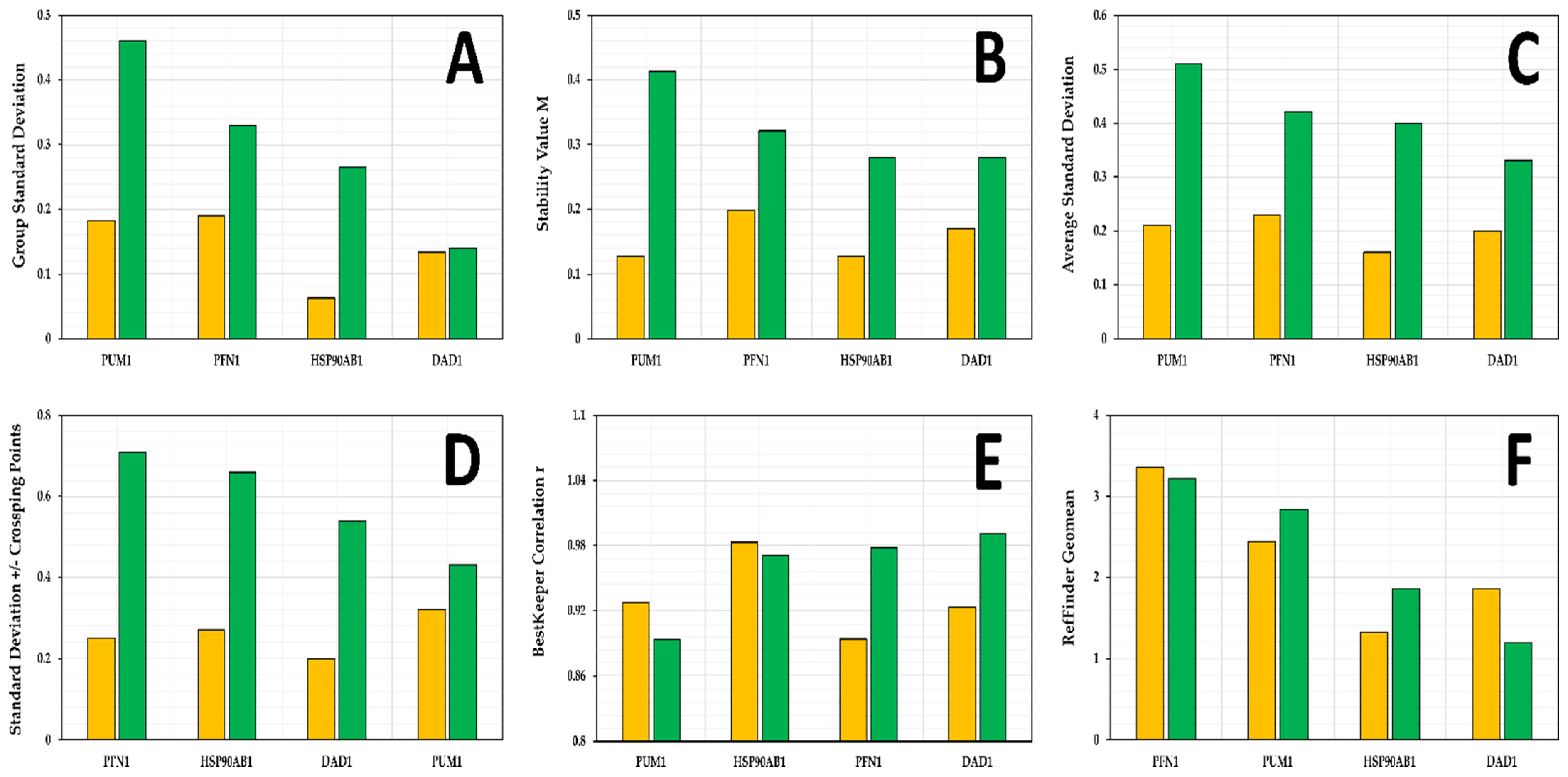
3.4. Candidate Reference Gene Stability in SK-BR-3 Cell Line
3.5. Candidate Reference Gene Stability in Replicate Cultures
3.6. Selection of Reference Genes for Further Validation
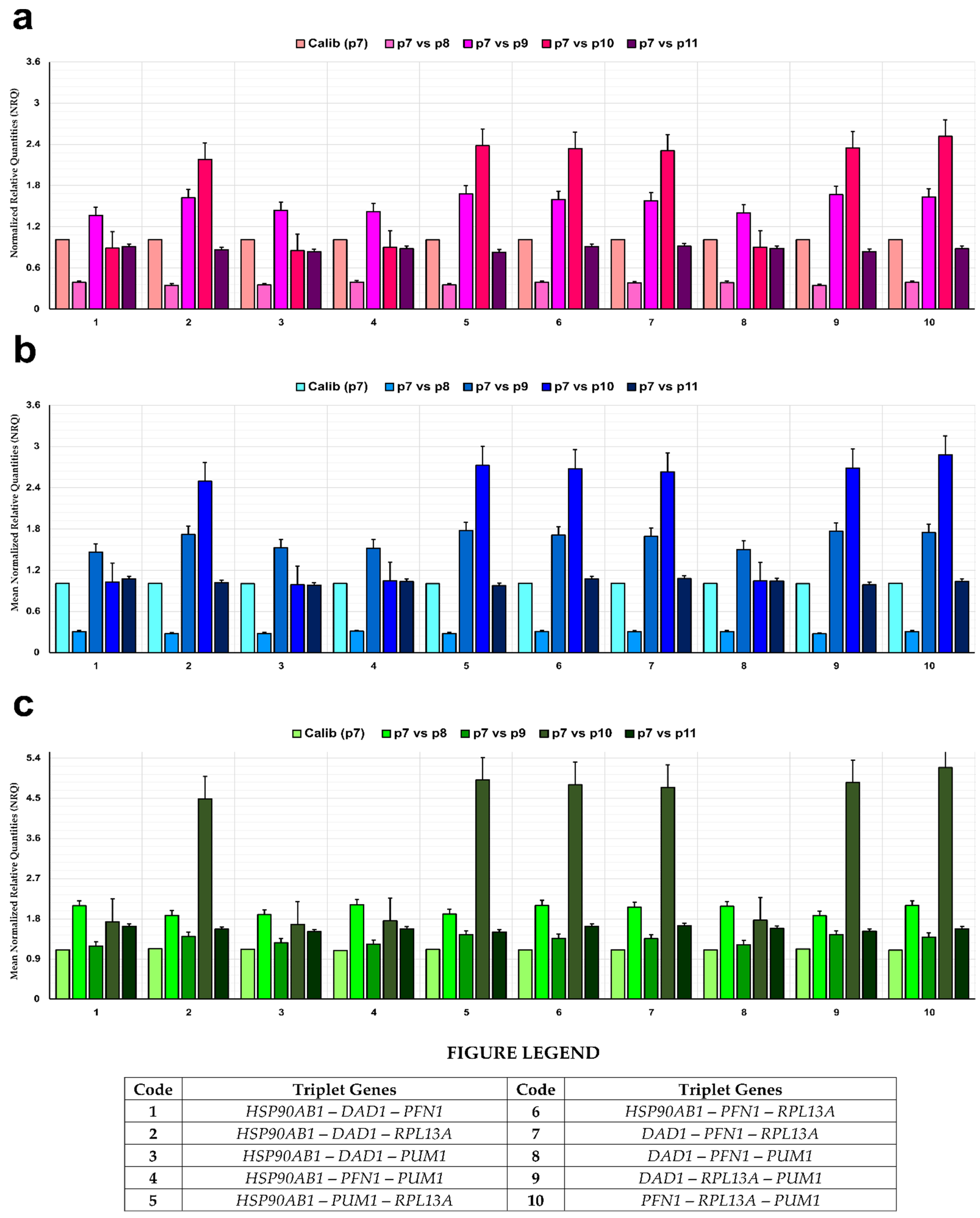
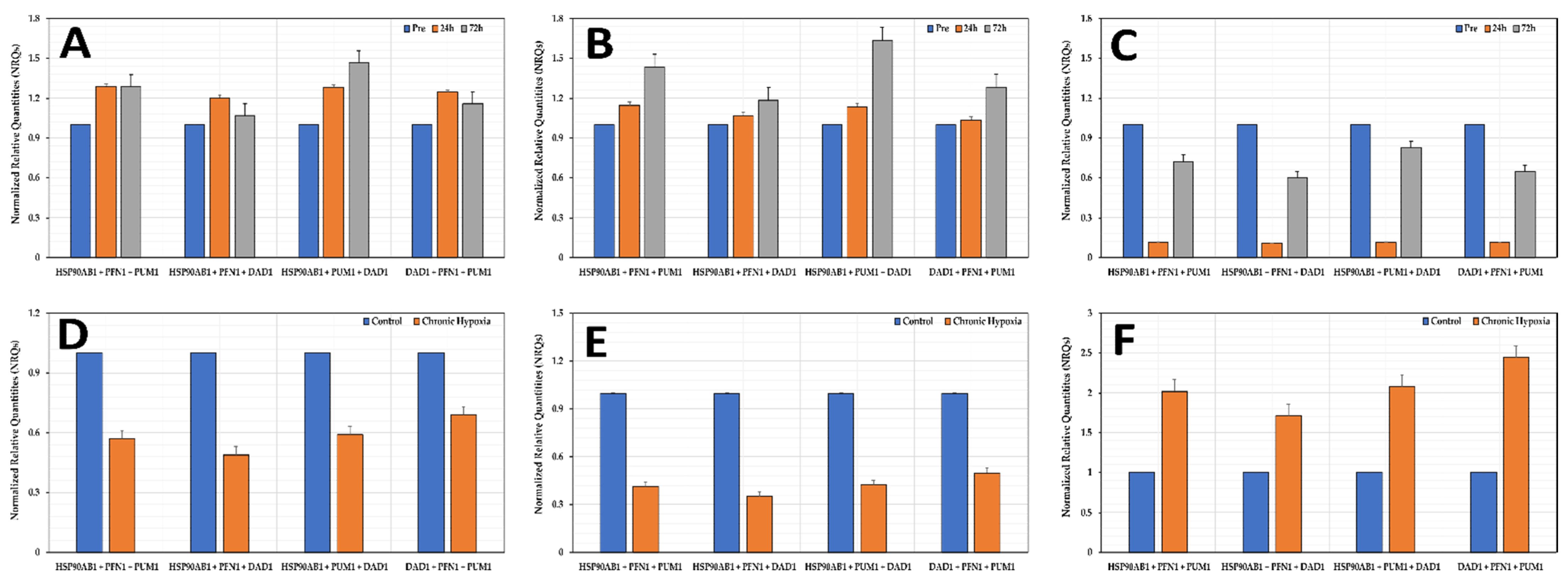
3.7. Normalization of Genes of Interest (GOIs)
3.8. Effects of Hypoxia on the Stability of the Selected Reference Genes
3.9. Experimental Validation of Reference Genes in Hypoxic Conditions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bustin, S.A. Real-time, fluorescence-based quantitative PCR: A snapshot of current procedures and preferences. Expert Rev. Mol. Diagn. 2005, 5, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Nolan, T. Pitfalls of Quantitative Real-Time Reverse-Transcription Polymerase Chain Reaction. J. Biomol. Tech. JBT 2004, 15, 155–166. [Google Scholar] [PubMed]
- Remans, T.; Keunen, E.; Bex, G.J.; Smeets, K.; Vangronsveld, J.; Cuypers, A. Reliable Gene Expression Analysis by Reverse Transcription-Quantitative PCR: Reporting and Minimizing the Uncertainty in Data Accuracy. Plant Cell 2014, 26, 3829–3837. [Google Scholar] [CrossRef] [PubMed]
- Kubista, M.; Andrade, J.M.; Bengtsson, M.; Forootan, A.; Jonak, J.; Lind, K.; Sindelka, R.; Sjöback, R.; Sjögreen, B.; Strömbom, L.; et al. The real-time polymerase chain reaction. Mol. Asp. Med. 2006, 27, 95–125. [Google Scholar] [CrossRef] [PubMed]
- Gabert, J.; Beillard, E.; Van Der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe Against Cancer Program. Leukemia 2003, 17, 2318–2357. [Google Scholar] [CrossRef] [PubMed]
- Wolffs, P.; Grage, H.; Hagberg, O.; Rådström, P. Impact of DNA Polymerases and Their Buffer Systems on Quantitative Real-Time PCR. J. Clin. Microbiol. 2004, 42, 408–411. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yeung, A.T.; Holloway, B.P.; Adams, P.S.; Shipley, G.L. Evaluation of dual-labeled fluorescent DNA probe purity versus performance in real-time PCR. BioTechniques 2004, 36, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Ginzinger, D.G. Gene quantification using real-time quantitative PCR: An emerging technology hits the mainstream. Exp. Hematol. 2002, 30, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Nolan, T.; Hands, R.E.; Bustin, S.A. Quantification of mRNA using real-time RT-PCR. Nat. Protoc. 2006, 1, 1559–1582. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.K.; Czechowski, T.; Scheible, W.-R. Eleven Golden Rules of Quantitative RT-PCR. Plant Cell 2008, 20, 1736–1737. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. qPCR: Quicker and easier but don′t be sloppy. Nat. Methods 2011, 8, 207–212. [Google Scholar] [CrossRef]
- Taylor, S.; Wakem, M.; Dijkman, G.; Alsarraj, M.; Nguyen, M. A practical approach to RT-qPCR—Publishing data that conform to the MIQE guidelines. Methods 2010, 50, S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Dheda, K.; Bustin, S.; Zumla, P.S.A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun. 2005, 6, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; Kubista, M.; Pfaffl, M.W. Reference gene validation software for improved normalization. In Real-Time PCR: Current Technology and Applications; Logan, J., Edwards, K., Saunders, N., Eds.; Caister Academic Press: Poole, UK, 2009; pp. 47–64. [Google Scholar]
- Bustin, S.A.; Benes, V.; Garson, J.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.; Shipley, G.; et al. The need for transparency and good practices in the qPCR literature. Nat. Methods 2013, 10, 1063–1067. [Google Scholar] [CrossRef] [PubMed]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef] [PubMed]
- Thellin, O.; Zorzi, W.; Lakaye, B.; De Borman, B.; Coumans, B.; Hennen, G.; Grisar, T.; Igout, A.; Heinen, E. Housekeeping genes as internal standards: Use and limits. J. Biotechnol. 1999, 75, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.D.; Sladek, R.; Greenwood, C.M.; Hudson, T.J. Control Genes and Variability: Absence of Ubiquitous Reference Transcripts in Diverse Mammalian Expression Studies. Genome Res. 2002, 12, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Barber, R.D.; Harmer, D.W.; Coleman, R.A.; Clark, B.J. GAPDH as a housekeeping gene: Analysis of GAPDH mRNA expression in a panel of 72 human tissues. Physiol. Genom. 2005, 21, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Ghani, M.; Sato, C.; Rogaeva, E. Segmental duplications in genome-wide significant loci and housekeeping genes; warning for GAPDH and ACTB. Neurobiol. Aging 2013, 34, 1710.e1–1710.e4. [Google Scholar] [CrossRef] [PubMed]
- Olsvik, P.A.; Søfteland, L.; Lie, K.K. Selection of reference genes for qRT-PCR examination of wild populations of Atlantic cod Gadus morhua. BMC Res. Notes 2008, 1, 47, Erratum in 2011, 4, 456. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; He, F.; Song, S.; Wang, J.; Yu, J. How many human genes can be defined as housekeeping with current expression data? BMC Genom. 2008, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Velculescu, V.E.; Madden, S.L.; Zhang, L.; Lash, A.E.; Yu, J.; Rago, C.; Lal, A.; Wang, C.J.; Beaudry, G.A.; Ciriello, K.M.; et al. Analysis of human transcriptomes. Nat. Genet. 1999, 23, 387–388. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, E.; Levanon, E.Y. Human housekeeping genes are compact. Trends Genet. 2003, 19, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Morse, D.L.; Carroll, D.; Weberg, L.; Borgstrom, M.C.; Ranger-Moore, J.; Gillies, R.J. Determining suitable internal standards for mRNA quantification of increasing cancer progression in human breast cells by real-time reverse transcriptase polymerase chain reaction. Anal. Biochem. 2005, 342, 69–77. [Google Scholar] [CrossRef] [PubMed]
- De Jonge, H.J.M.; Fehrmann, R.; De Bont, E.S.J.M.; Hofstra, R.; Gerbens, F.; Kamps, W.A.; de Vries, E.; Van Der Zee, A.G.J.; Meerman, G.J.T.; Ter Elst, A. Evidence Based Selection of Housekeeping Genes. PLoS ONE 2007, 2, e898. [Google Scholar] [CrossRef] [PubMed]
- Lyng, M.B.; Laenkholm, A.-V.; Pallisgaard, N.; Ditzel, H.J. Identification of genes for normalization of real-time RT-PCR data in breast carcinomas. BMC Cancer 2008, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Gur-Dedeoglu, B.; Konu, O.; Bozkurt, B.; Ergul, G.; Seckin, S.; Yulug, I.G. Identification of Endogenous Reference Genes for qRT-PCR Analysis in Normal Matched Breast Tumor Tissues. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2009, 17, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Avnet, S.; Salerno, M.; Chano, T.; Baldini, N. Identification and Validation of Housekeeping Genes for Gene Expression Analysis of Cancer Stem Cells. PLoS ONE 2016, 11, e0149481. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Guertler, R.; Naim, S.; Nixdorf, S.; Fedier, A.; Hacker, N.F.; Heinzelmann-Schwarz, V. Careful Selection of Reference Genes Is Required for Reliable Performance of RT-qPCR in Human Normal and Cancer Cell Lines. PLoS ONE 2013, 8, e59180. [Google Scholar] [CrossRef] [PubMed]
- Maltseva, D.V.; Khaustova, N.A.; Fedotov, N.N.; Matveeva, E.O.; Lebedev, A.E.; Shkurnikov, M.U.; Galatenko, V.V.; Schumacher, U.; Tonevitsky, A.G. High-throughput identification of reference genes for research and clinical RT-qPCR analysis of breast cancer samples. J. Clin. Bioinform. 2013, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Kılıç, Y.; Çelebiler, A.; Sakızlı, M. Selecting housekeeping genes as references for the normalization of quantitative PCR data in breast cancer. Clin. Transl. Oncol. 2013, 16, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.-L.; Zhao, H.; Ma, T.-F.; Ge, F.; Chen, C.-S.; Zhang, Y.-P. Identification of Valid Reference Genes for the Normalization of RT-qPCR Expression Studies in Human Breast Cancer Cell Lines Treated with and without Transient Transfection. PLoS ONE 2015, 10, e0117058. [Google Scholar] [CrossRef] [PubMed]
- Tilli, T.M.; Castro, C.D.S.; Tuszynski, J.A.; Carels, N. A strategy to identify housekeeping genes suitable for analysis in breast cancer diseases. BMC Genom. 2016, 17, 639. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, A.P.; Balmana, M.; Reis, C.A.; Beltrao, E.I. Identification of appropriate housekeeping genes for quantitative RT-PCR analysis in MDA-MB-231 and NCI-H460 human cancer cell lines under hypoxia and serum deprivation. J. Mol. Clin. Med. 2018, 1, 127–134. [Google Scholar] [CrossRef]
- Jo, J.; Choi, S.; Oh, J.; Lee, S.-G.; Choi, S.Y.; Kim, K.K.; Park, C. Conventionally used reference genes are not outstanding for normalization of gene expression in human cancer research. BMC Bioinform. 2019, 20 (Suppl. S10), 245. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Kudryavtseva, A.V.; Snezhkina, A.V.; Lakunina, V.A.; Beniaminov, A.D.; Melnikova, N.V.; Dmitriev, A.A. Pan-Cancer Analysis of TCGA Data Revealed Promising Reference Genes for qPCR Normalization. Front. Genet. 2019, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in 2020, 70, 313. [Google Scholar] [CrossRef] [PubMed]
- Heer, E.; Harper, A.; Escandor, N.; Sung, H.; McCormack, V.; Fidler-Benaoudia, M.M. Global burden and trends in premenopausal and postmenopausal breast cancer: A population-based study. Lancet Glob. Health 2020, 8, e1027–e1037. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised Risk Predictor of Breast Cancer Based on Intrinsic Subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Dowsett, M.; Sestak, I.; Knowles, E.L.; Sidhu, K.; Dunbier, A.; Cowens, J.W.; Ferree, S.; Storhoff, J.; Schaper, C.; Cuzick, J. Comparison of PAM50 Risk of Recurrence Score With Oncotype DX and IHC4 for Predicting Risk of Distant Recurrence After Endocrine Therapy. J. Clin. Oncol. 2013, 31, 2783–2790. [Google Scholar] [CrossRef] [PubMed]
- Gnant, M.; Filipits, M.; Greil, R.; Stoeger, H.; Rudas, M.; Bago-Horvath, Z.; Mlineritsch, B.; Kwasny, W.; Knauer, M.; Singer, C.; et al. Predicting distant recurrence in receptor-positive breast cancer patients with limited clinicopathological risk: Using the PAM50 Risk of Recurrence score in 1478 postmenopausal patients of the ABCSG-8 trial treated with adjuvant endocrine therapy alone. Ann. Oncol. 2013, 25, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Vincent-Salomon, A.; Pivot, X.; Sertier, A.-S.; Thomas, E.; Tonon, L.; Boyault, S.; Mulugeta, E.; Treilleux, I.; MacGrogan, G.; et al. A whole-genome sequence and transcriptome perspective on HER2-positive breast cancers. Nat. Commun. 2016, 7, 12222. [Google Scholar] [CrossRef] [PubMed]
- Cejalvo, J.M.; Pascual, T.; Fernandez-Martinez, A.; Maristany, F.B.; Gomis, R.; Perou, C.; Munoz, M.; Prat, A. Clinical implications of the non-luminal intrinsic subtypes in hormone receptor-positive breast cancer. Cancer Treat. Rev. 2018, 67, 63–70. [Google Scholar] [CrossRef] [PubMed]
- SK-BR-3: Human Breast Cancer Cell Line (ATCC HTB-30) Memorial Sloan Kettering Cancer Center. Available online: https://www.mskcc.org/research-advantage/support/technology/tangible-material/human-breast-cell-line-sk-br-3 (accessed on 30 November 2020).
- Kenny, P.A.; Lee, G.Y.; Myers, C.A.; Neve, R.M.; Semeiks, J.R.; Spellman, P.T.; Lorenz, K.; Lee, E.H.; Barcellos-Hoff, M.H.; Petersen, O.W.; et al. The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Mol. Oncol. 2007, 1, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.-L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular Profiling of Breast Cancer Cell Lines Defines Relevant Tumor Models and Provides a Resource for Cancer Gene Discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, R.A.; Harvey, K.A.; Walker, C.; Altenburg, J.; Xu, Z.; Terry, C.; Camarillo, I.; Jones-Hall, Y.; Mariash, C. Characterization of synergistic anti-cancer effects of docosahexaenoic acid and curcumin on DMBA-induced mammary tumorigenesis in mice. BMC Cancer 2013, 13, 418. [Google Scholar] [CrossRef] [PubMed]
- Maristany, F.B.; Griguolo, G.; Pascual, T.; Pare, L.; Nuciforo, P.; Llombart-Cussac, A.; Bermejo, B.; Oliveira, M.; Morales, S.; Martínez, N.; et al. Phenotypic changes of HER2-positive breast cancer during and after dual HER2 blockade. Nat. Commun. 2020, 11, 385. [Google Scholar] [CrossRef] [PubMed]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Triner, D.; Shah, Y.M. Hypoxia-inducible factors: A central link between inflammation and cancer. J. Clin. Investig. 2016, 126, 3689–3698. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Wang, M.; Schmid, T.; Xin, Z.; Kozhuharova, L.; Yu, W.-K.; Huang, Y.; Cai, F.; Biskup, E. Hypoxia in Breast Cancer—Scientific Translation to Therapeutic and Diagnostic Clinical Applications. Front. Oncol. 2021, 11, 652266. [Google Scholar] [CrossRef] [PubMed]
- Denko, N.C.; Fontana, L.A.; Hudson, K.M.; Sutphin, P.D.; Raychaudhuri, S.; Altman, R.; Giaccia, A.J. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene 2003, 22, 5907–5914. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2015, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.C.; Colaprico, A.; Olsen, C.; D′Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. F1000Research 2016, 5, 1542. [Google Scholar] [CrossRef] [PubMed]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium; Carbon, S.; Douglass, E.; Good, B.M.; Unni, D.R.; Harris, N.L.; Mungall, C.J.; Basu, S.; Chisholm, R.L.; Dodson, R.J.; et al. The Gene Ontology resource: Enriching a Gold mine. Nucleic Acids Res. 2020, 49, D325–D334. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Dalmer, T.R.A.; Clugston, R.D. Gene ontology enrichment analysis of congenital diaphragmatic hernia-associated genes. Pediatr. Res. 2018, 85, 13–19, Erratum in 2019, 86, 676. [Google Scholar] [CrossRef] [PubMed]
- Pirsko, V.; Cakstina, I.; Priedite, M.; Dortane, R.; Feldmane, L.; Nakazawa-Miklasevica, M.; Daneberga, Z.; Gardovskis, J.; Miklasevics, E. An Effect of Culture Media on Epithelial Differentiation Markers in Breast Cancer Cell Lines MCF7, MDA-MB-436 and SkBr3. Medicina 2018, 54, 11. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Nitisa, D.; Pirsko, V.; Cakstina, I. Selecting suitable reference genes for qPCR normalization: A comprehensive analysis in MCF-7 breast cancer cell line. BMC Mol. Cell Biol. 2020, 21, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of Real-Time Quantitative Reverse Transcription-PCR Data: A Model-Based Variance Estimation Approach to Identify Genes Suited for Normalization, Applied to Bladder and Colon Cancer Data Sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper—Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Xiao, P.; Chen, D.; Xu, L.; Zhang, B. miRDeepFinder: A miRNA analysis tool for deep sequencing of plant small RNAs. Plant. Mol. Biol. 2012, 80, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, L.; Li, R.; Zhang, M.; Li, Y.; Wang, H.; Wang, S.; Bao, Z. Systematic identification and validation of the reference genes from 60 RNA-Seq libraries in the scallop Mizuhopecten yessoensis. BMC Genom. 2019, 20, 288. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, J.; Mortier, G.; De Paepe, A.; Speleman, F.; Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007, 8, R19. [Google Scholar] [CrossRef] [PubMed]
- D′haene, B.; Hellemans, J. The importance of quality control during qPCR data analyis. Int. Drug Discov. 2010, 18, 24. [Google Scholar]
- Cooper, L.A.; Demicco, E.G.; Saltz, J.H.; Powell, R.T.; Rao, A.; Lazar, A.J. PanCancer insights from The Cancer Genome Atlas: The pathologist′s perspective. J. Pathol. 2017, 244, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Velázquez, M.; Ostoa-Saloma, P.; Palacios-Arreola, M.I.; Castro, K.E.N.; Castro, J.I.; Morales-Montor, J. The Role of Cytokines in Breast Cancer Development and Progression. J. Interf. Cytokine Res. 2015, 35, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A. TGF-ß signaling, tumor microenvironment and tumor progression: The butterfly effect. Front. Biosci. 2010, 15, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Vincent, T.; Neve, E.P.A.; Johnson, J.R.; Kukalev, A.; Rojo, F.; Albanell, J.; Pietras, K.; Virtanen, I.; Philipson, L.; Leopold, P.L.; et al. A SNAIL1–SMAD3/4 transcriptional repressor complex promotes TGF-β mediated epithelial–mesenchymal transition. Nature 2009, 11, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Honma, S.; Shimodaira, K.; Shimizu, Y.; Tsuchiya, N.; Saito, H.; Yanaihara, T.; Okai, T. The Influence of Inflammatory Cytokines on Estrogen Production and Cell Proliferation in Human Breast Cancer Cells. Endocr. J. 2002, 49, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, K.; Mishima, K.; Ohsawa, M.; Aozasa, K. Carcinoma of stomach and breast with lymphoid stroma: Localisation of Epstein-Barr virus. J. Clin. Pathol. 1994, 47, 538–540. [Google Scholar] [CrossRef] [PubMed]
- Di Lonardo, A.; Venuti, A.; Marcante, M.L. Human papillomavirus in breast cancer. Breast Cancer Res. Treat. 1992, 21, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Buehring, G.C.; Shen, H.M.; Jensen, H.M.; Block, G. Bovine leukemia virus infection is significantly associated with risk of breast cancer. Proc. Am. Assoc. Cancer Res. 2007, 48, 1747. [Google Scholar]
- Wang, Y.; Holland, J.F.; Bleiweiss, I.J.; Melana, S.; Liu, X.; Pelisson, I.; Cantarella, A.; Stellrecht, K.; Mani, S.; Pogo, B.G. Detection of mammary tumor virus env gene-like sequences in human breast cancer. Cancer Res. 1995, 55, 5173–5179. [Google Scholar] [PubMed]
- Lawson, J.S.; Glenn, W.K.; Heng, B.; Ye, Y.; Tran, B.; Lutze-Mann, L.; Whitaker, N.J. Koilocytes indicate a role for human papilloma virus in breast cancer. Br. J. Cancer 2009, 101, 1351–1356. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bian, L.; Zhao, L.; Dong, Z.; Gao, X.; Luan, H.; Sun, Y.; Song, H. Identification of genes for normalization of quantitative real-time PCR data in ovarian tissues. Acta Biochim. et Biophys. Sin. (Shanghai) 2010, 42, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.J.; Rios, L.; Trivedi, P.; D′Souza, K.; Cowie, A.; Nzirorera, C.; Webster, D.; Brunt, K.; Legare, J.-F.; Hassan, A.; et al. Validation of optimal reference genes for quantitative real time PCR in muscle and adipose tissue for obesity and diabetes research. Sci. Rep. 2017, 7, 3612. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, J.; Słowińska, M.; Cierniak, P.; Kopcewicz, M.; Walendzik, K.; Frazier, T.; Gawrońska-Kozak, B. The effect of hypoxia on the proteomic signature of pig adipose-derived stromal/stem cells (pASCs). Sci. Rep. 2020, 10, 20035. [Google Scholar] [CrossRef] [PubMed]
- Sonna, L.A.; Cullivan, M.L.; Sheldon, H.K.; Pratt, R.E.; Lilly, C.M. Effect of hypoxia on gene expression by human hepatocytes (HepG2). Physiol. Genom. 2003, 12, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Sanjay, A.; Fu, J.; Kreibich, G. DAD1 Is Required for the Function and the Structural Integrity of the Oligosaccharyltransferase Complex. J. Biol. Chem. 1998, 273, 26094–26099. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T.; Sekiguchi, T.; Kuraoka, A.; Fukushima, K.; Shibata, Y.; Komiyama, S.; Nishimoto, T. Molecular cloning of a human cDNA encoding a novel protein, DAD1, whose defect causes apoptotic cell death in hamster BHK21 cells. Mol. Cell. Biol. 1993, 13, 6367–6374. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.; Collins, P.G.; Kelleher, D.J.; Gilmore, R. The essential OST2 gene encodes the 16-kD subunit of the yeast oligosaccharyltransferase, a highly conserved protein expressed in diverse eukaryotic organisms. J. Cell Biol. 1995, 131, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kondoh, N.; Shuda, M.; Matsubara, O.; Imazeki, N.; Ryo, A.; Wakatsuki, T.; Hada, A.; Goseki, N.; Igari, T.; et al. Enhanced expression of mRNAs of antisecretory factor-1, gp96, DAD1 and CDC34 in human hepatocellular carcinomas. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2001, 1536, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Hoang, T.; Saeed, B.; Ng, S.C. Induction of apoptosis in prostatic tumor cell line DU145 by staurosporine, a potent inhibitor of protein kinases. Prostate 1996, 29, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Hao, P.; Dutta, B.; Cheow, E.S.H.; Sim, K.H.; Gan, C.S.; Lim, S.K.; Sze, S.K. Hypoxia Modulates A431 Cellular Pathways Association to Tumor Radioresistance and Enhanced Migration Revealed by Comprehensive Proteomic and Functional Studies. Mol. Cell. Proteom. 2013, 12, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Bae, Y.H.; Roy, P. Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adhes. Migr. 2012, 6, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Witke, W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004, 14, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.H.; Ding, Z.; Zou, L.; Wells, A.; Gertler, F.; Roy, P. Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J. Cell. Physiol. 2008, 219, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Das, T.; Bae, Y.H.; Wells, A.; Roy, P. Profilin-1 overexpression upregulates PTEN and suppresses AKT activation in breast cancer cells. J. Cell. Physiol. 2008, 218, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Ji, S.; Qin, Y.; Yang, J.; Xu, J.; Zhang, B.; Xu, W.; Liu, J.; Shi, S.; Liu, L.; et al. Profilin-1 suppresses tumorigenicity in pancreatic cancer through regulation of the SIRT3-HIF1α axis. Mol. Cancer 2014, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Gau, D.; Bae, Y.; Ohayon, L. Breast cancer cell invasiveness is stimulated by loss of membrane interaction of actinbinding protein profilin1 via altered phosphoinositide metabolism. FASEB J. 2019, 33, 488.13. [Google Scholar] [CrossRef]
- Guan, X.; Chen, S.; Liu, Y.; Wang, L.-L.; Zhao, Y.; Zong, Z.-H. PUM1 promotes ovarian cancer proliferation, migration and invasion. Biochem. Biophys. Res. Commun. 2018, 497, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, S.J.; Han, S.M.; Paik, S.Y.; Hur, S.Y.; Kim, Y.W.; Lee, J.M.; Namkoong, S.E. Increased Glyceraldehyde-3-Phosphate Dehydrogenase Gene Expression in Human Cervical Cancers. Gynecol. Oncol. 1998, 71, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Rondinelli, R.H.; Epner, D.E.; Tricoli, J.V. Increased glyceraldehyde-3-phosphate dehydrogenase gene expression in late pathological stage human prostate cancer. Prostate Cancer Prostatic Dis. 1997, 1, 66–72. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Source | Selected Candidate Reference Genes |
|---|---|---|
| Breast cancer various cell lines | Literature | ACTB [26,29,32,34], CCSER2 [35], GAPDH [34], HNRNPL [37], HSP90AB1 [31], PCBP1 [37], PGK1 [30], PPIA [36], PUM1 [28,33,35], RNA18S [26,34], RNA28S [66], RPL13A [27,33], SF3A1 [32,38] |
| HER2-E tissue samples | TCGA (Novel) | BSG, CFL1, DAD1, EIF5A, GABARAP, NACA, PFN1, PSMB4, RBX1, TPT1, TUBA1B, UBC |
| Genes of Interest (GOI) | Expression Atlas | AURKA, BUB1, SNAI1 |
| GO ID | GO Term | No. of Genes * | Fold Enrichment | Raw p Value | FDR |
|---|---|---|---|---|---|
| GO:0044003 | Modulation by symbiont of host process | 3 | 46.10 | 4.24 × 10−05 | 3.05 × 10−02 |
| GO:0006090 | Pyruvate metabolic process | 3 | 38.31 | 7.20 × 10−05 | 3.68 × 10−02 |
| GO:0061418 | Regulation of transcription from RNA polymerase II promoter in response to hypoxia | 3 | 34.87 | 9.43 × 10−05 | 4.27 × 10−02 |
| GO:0048524 | Positive regulation of viral process | 3 | 32.67 | 7.14 × 10−06 | 1.03 × 10−02 |
| GO:0019058 | Viral life cycle | 5 | 20.33 | 4.26 × 10−06 | 7.52 × 10−03 |
| GO:0006417 | Regulation of translation | 5 | 12.12 | 4.90 × 10−05 | 3.26 × 10−02 |
| GO:0071345 | Cellular response to cytokine stimulus | 9 | 7.86 | 8.38 × 10−07 | 3.33 × 10−03 |
| GO:0043066 | Negative regulation of apoptotic process | 7 | 6.96 | 4.13 × 10−05 | 3.12 × 10−02 |
| GO:0006139 | Nucleobase containing compound metabolic process | 11 | 3.64 | 6.05 × 10−05 | 3.56 × 10−02 |
| GO:0010604 | Positive regulation of macromolecule metabolic process | 12 | 3.05 | 1.33 × 10−04 | 5.02 × 10−02 |
| GO:0043170 | Macromolecule metabolic process | 17 | 2.43 | 2.16 × 10−05 | 2.01 × 10−02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, N.; Mitre, I.; Nitisa, D.; Pirsko, V.; Cakstina-Dzerve, I. Identification of Novel Endogenous Controls for qPCR Normalization in SK-BR-3 Breast Cancer Cell Line. Genes 2021, 12, 1631. https://doi.org/10.3390/genes12101631
Jain N, Mitre I, Nitisa D, Pirsko V, Cakstina-Dzerve I. Identification of Novel Endogenous Controls for qPCR Normalization in SK-BR-3 Breast Cancer Cell Line. Genes. 2021; 12(10):1631. https://doi.org/10.3390/genes12101631
Chicago/Turabian StyleJain, Nityanand, Ingrida Mitre, Dina Nitisa, Valdis Pirsko, and Inese Cakstina-Dzerve. 2021. "Identification of Novel Endogenous Controls for qPCR Normalization in SK-BR-3 Breast Cancer Cell Line" Genes 12, no. 10: 1631. https://doi.org/10.3390/genes12101631
APA StyleJain, N., Mitre, I., Nitisa, D., Pirsko, V., & Cakstina-Dzerve, I. (2021). Identification of Novel Endogenous Controls for qPCR Normalization in SK-BR-3 Breast Cancer Cell Line. Genes, 12(10), 1631. https://doi.org/10.3390/genes12101631