
Comparative RNA-Seq Analyses of Solenopsis japonica (Hymenoptera: Formicidae) Reveal Gene in Response to Cold Stress


Abstract
:
1. Introduction
2. Materials and Methods
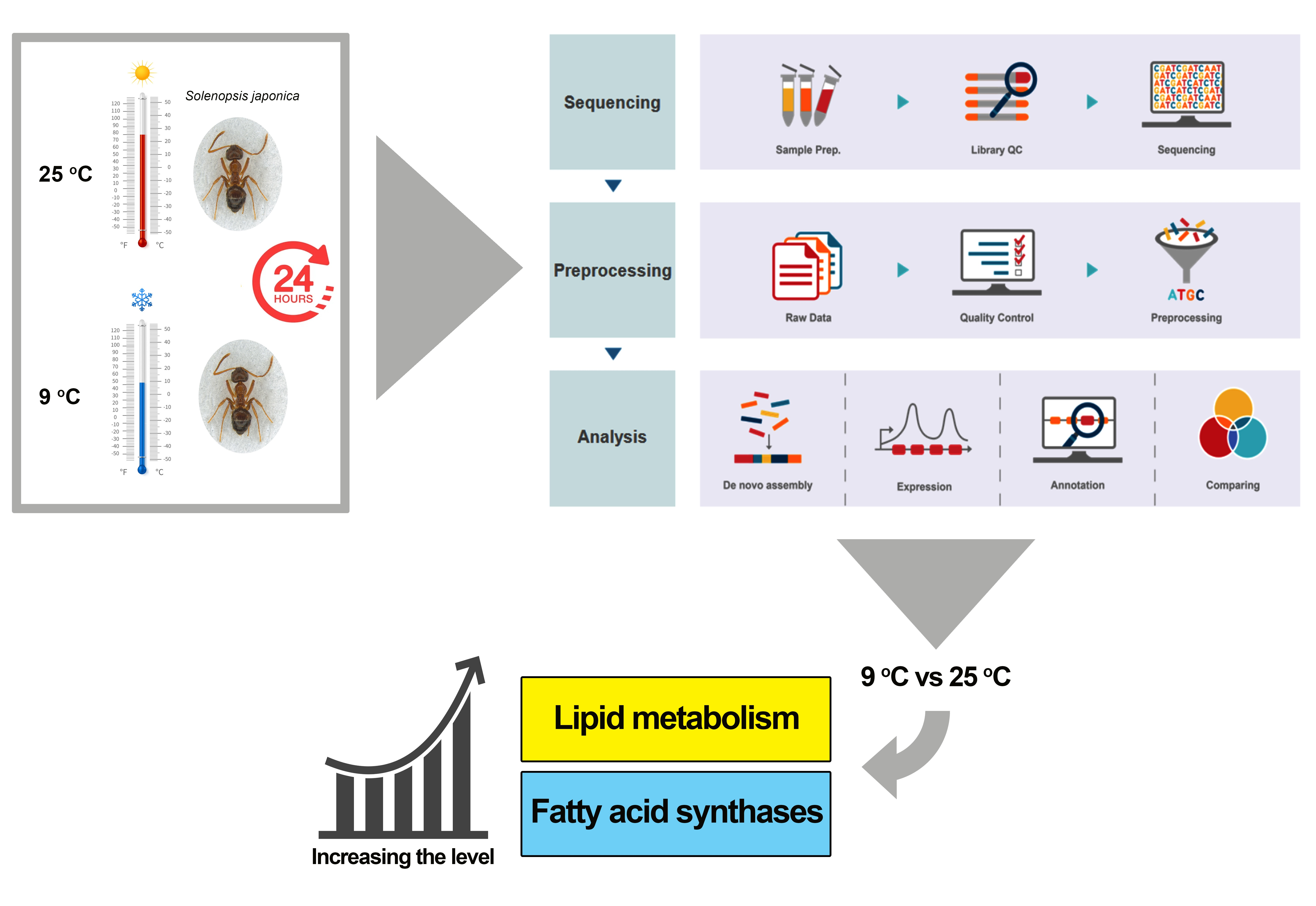
2.1. Insect Rearing, Exposure Temperatures and Sample Preparation
2.2. RNA Extraction and RT-qPCR
2.3. Illumina Sequencing
2.4. De Novo Assembly
2.5. Gene Functional Annotation
2.6. Differential Gene Expression Analysis
2.7. Quantitative RT-PCR Validation
2.8. Data Analysis
3. Results
3.1. Sequencing, RNA-Seq Assembly, and Functional Annotation
3.2. Annotation of Predicted Proteins
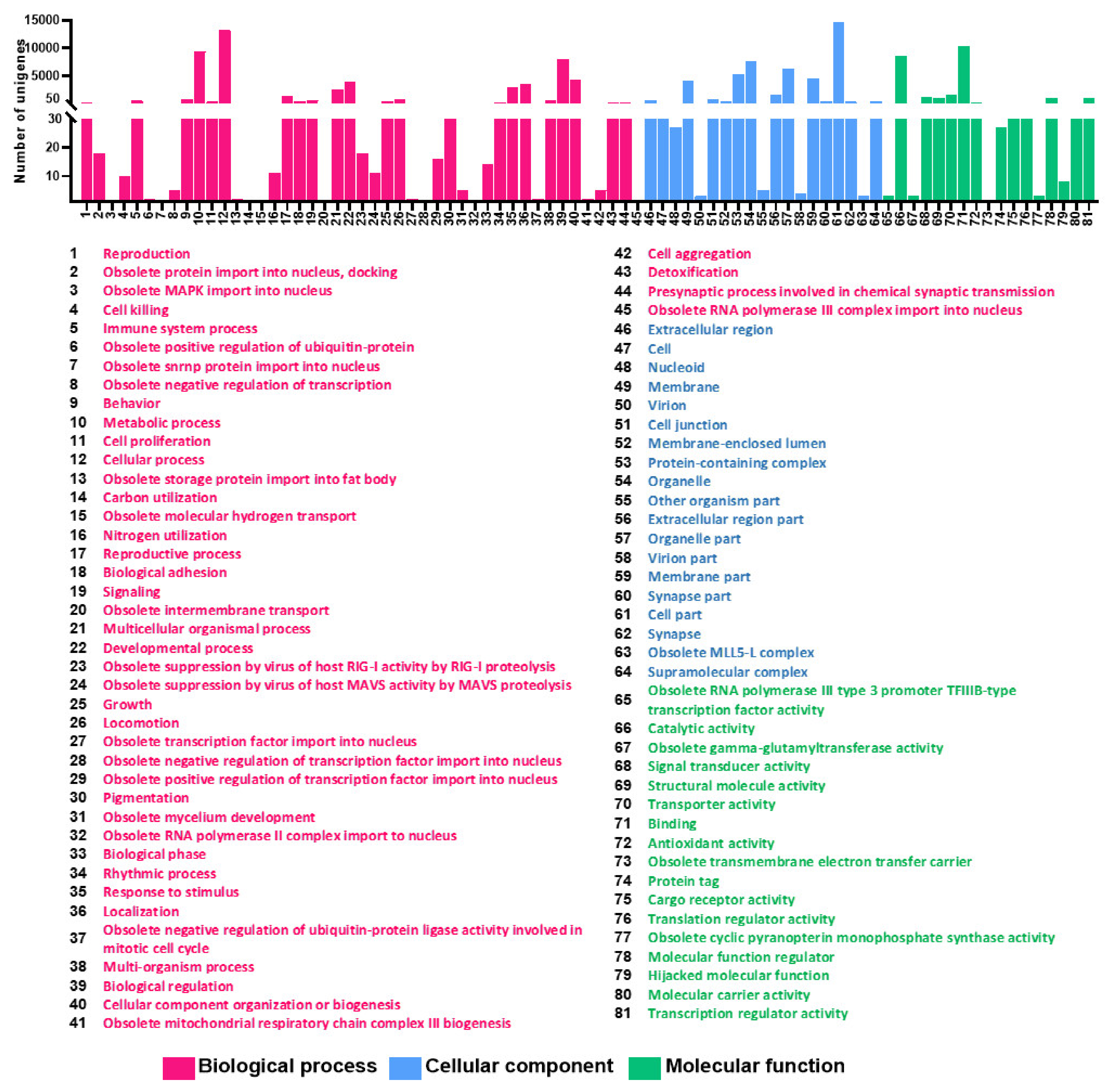
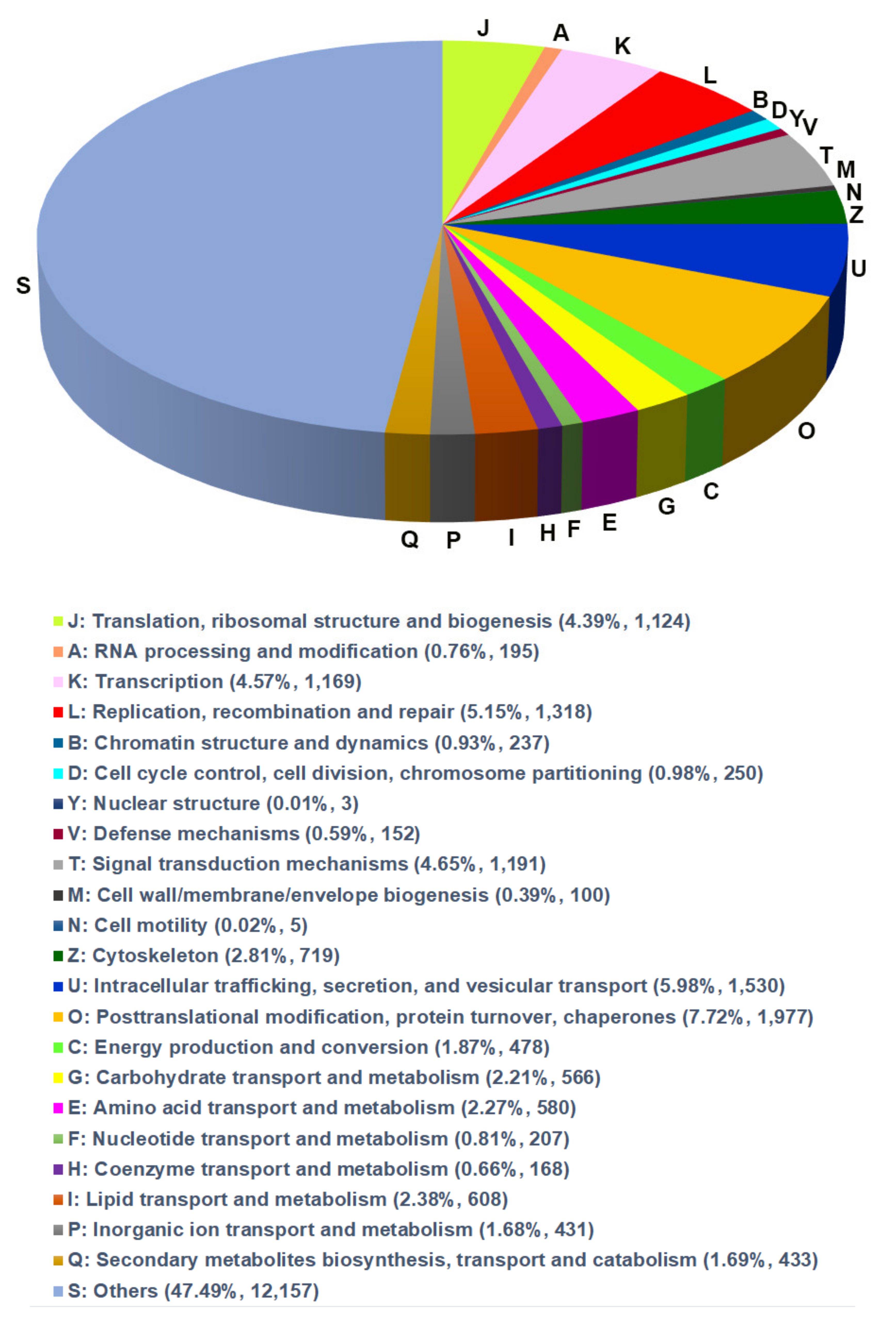
3.3. GO and EggNOG Analysis for Global Functional Classification
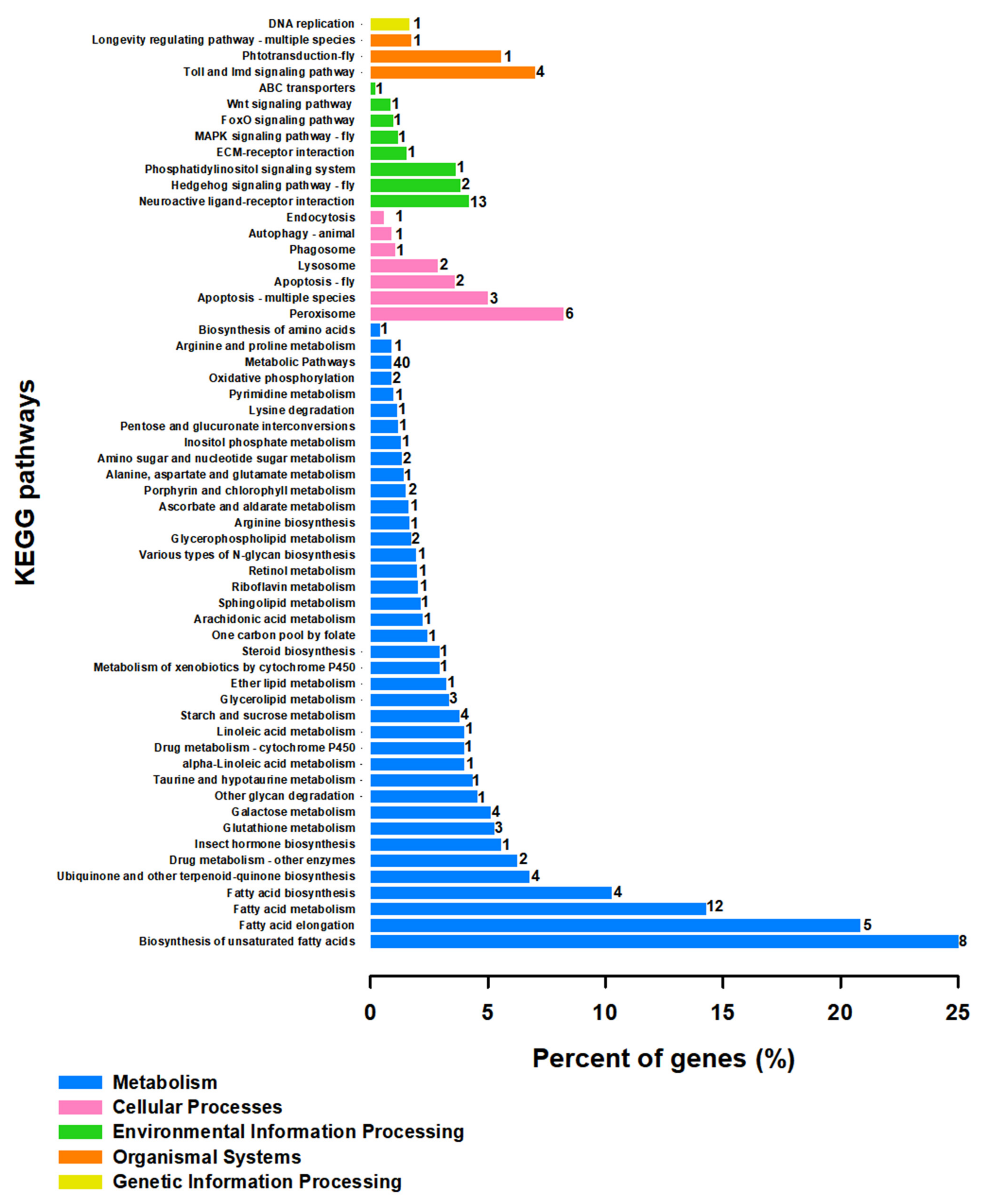
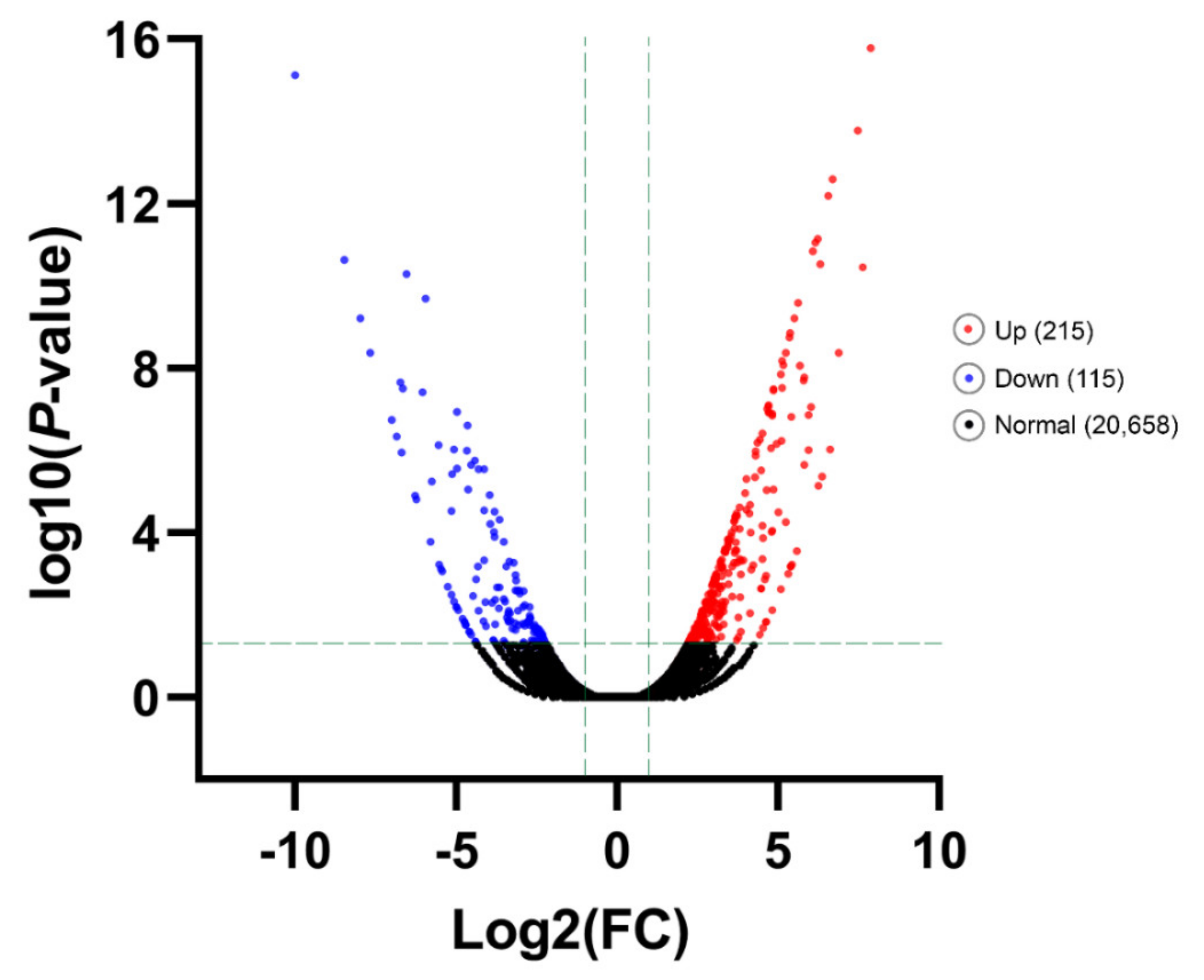
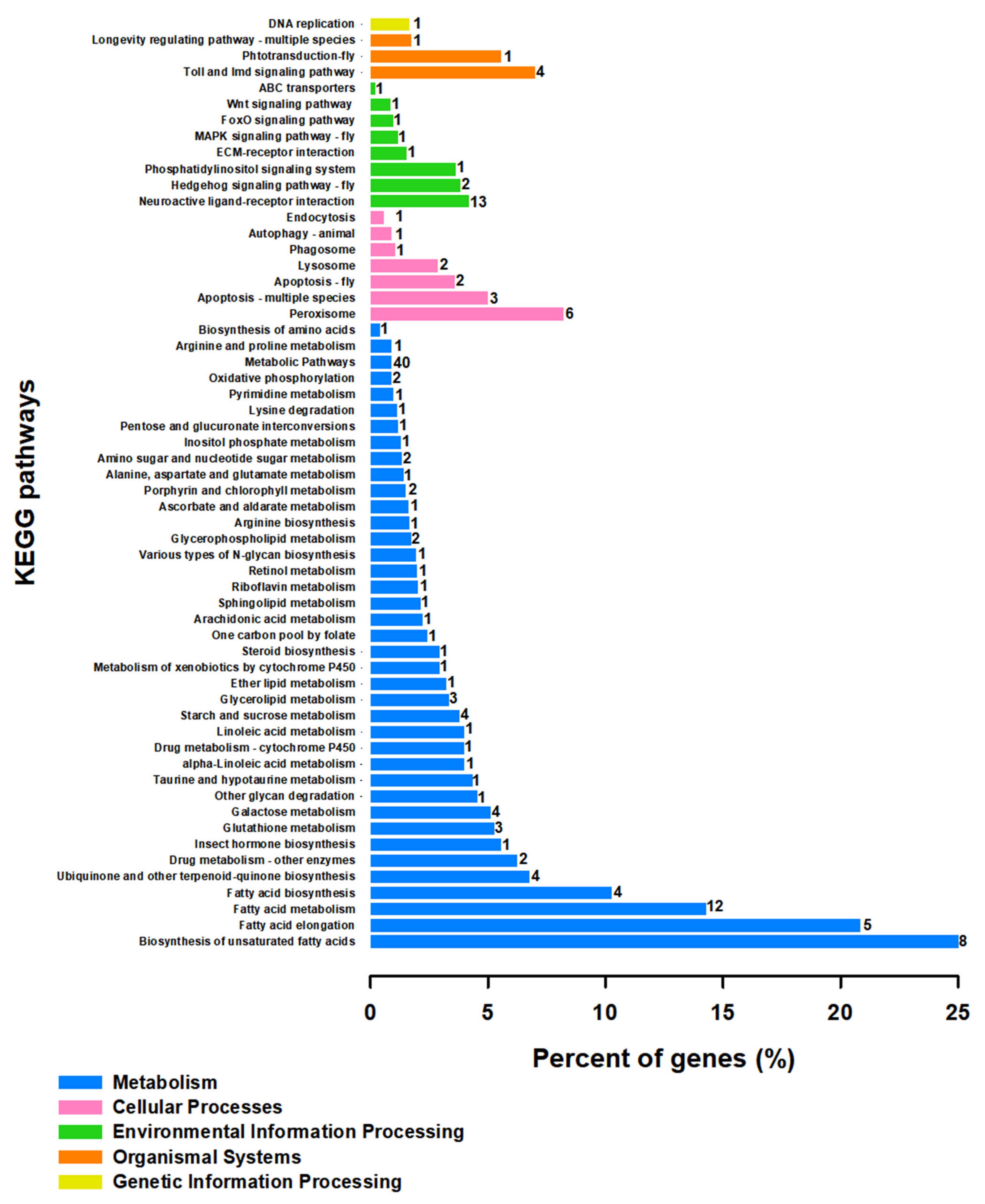
3.4. Differentially Gene Expression under Different Temperature
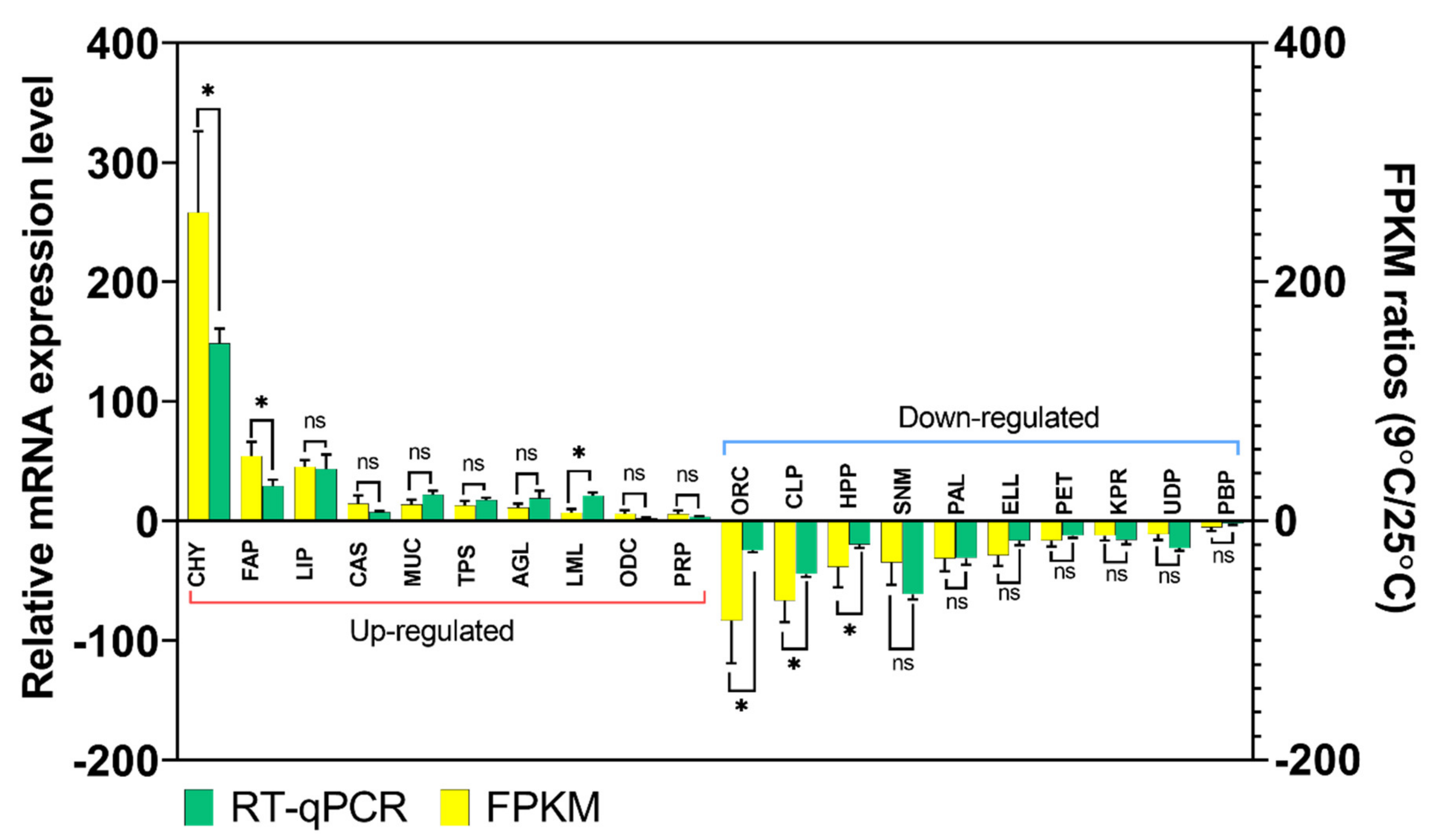
3.5. Validation of Gene Expression Profiles by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Briano, J.; Calcaterra, L.; Varone, L. Fire ants (Solenopsis spp.) and their natural enemies in southern South America. Psyche 2012, 2012, 1980–1984. [Google Scholar]
- Yamaguchi, T.; Hasegawa, M. An experiment on ant predation in soil using a new bait trap method. Ecol. Res. 1996, 11, 11–16. [Google Scholar] [CrossRef]
- Teranishi, C. Japanese ants, their behavior and distribution (1). Zool. Mag. 1929, 41, 239–251. [Google Scholar]
- Brown, V.K.; Gange, A.C. Insect herbivory insect below ground. In Advances in Ecological Research; Elsevier: Amsterdam, The Netherlands, 1990; Volume 20, pp. 1–58. ISBN 0065-2504. [Google Scholar]
- Cramer, W.; Bondeau, A.; Woodward, F.I.; Prentice, I.C.; Betts, R.A.; Brovkin, V.; Cox, P.M.; Fisher, V.; Foley, J.A.; Friend, A.D. Global response of terrestrial ecosystem structure and function to CO2 and climate change: Results from six dynamic global vegetation models. Glob. Chang. Biol. 2001, 7, 357–373. [Google Scholar] [CrossRef] [Green Version]
- Tylianakis, J.M.; Didham, R.K.; Bascompte, J.; Wardle, D.A. Global change and species interactions in terrestrial ecosystems. Ecol. Lett. 2008, 11, 1351–1363. [Google Scholar] [CrossRef]
- Li, H.; Zhao, X.; Qiao, H.; He, X.; Tan, J.; Hao, D. Comparative transcriptome analysis of the heat stress response in Monochamus alternatus Hope (Coleoptera: Cerambycidae). Front. Physiol. 2020, 10, 1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrad, R.; Booth, D.T.; Furlong, M.J. The effect of rearing temperature on development, body size, energetics and fecundity of the diamondback moth. Bull. Entomol. Res. 2016, 106, 175. [Google Scholar] [CrossRef]
- Bale, J.S. Insects and low temperatures: From molecular biology to distributions and abundance. Philos. Trans. R. Soc. Lond. Ser. B. Biol. Sci. 2002, 357, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.E. Principles of insect low temperature tolerance. In Insects at Low Temperature; Springer: Berlin/Heidelberg, Germany, 1991; pp. 17–46. [Google Scholar]
- Turnock, W.J.; Lamb, R.J.; Bodnaryk, R.P. Effects of cold stress during pupal diapause on the survival and development of Mamestra configurata (Lepidoptera: Noctuidae). Oecologia 1983, 56, 185–192. [Google Scholar] [CrossRef]
- Hutchinson, L.A.; Bale, J.S. Effects of sublethal cold stress on the aphid Rhopalosiphum padi. J. Appl. Ecol. 1994, 31, 102–108. [Google Scholar] [CrossRef]
- Bale, J.S. Classes of insect cold hardiness. Funct. Ecol. 1993, 7, 751–753. [Google Scholar]
- Gillooly, J.F.; Brown, J.H.; West, G.B.; Savage, V.M.; Charnov, E.L. Effects of size and temperature on metabolic rate. Science 2001, 293, 2248–2251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rall, B.C.; Vucic-Pestic, O.; Ehnes, R.B.; Emmerson, M.; Brose, U. Temperature, predator–prey interaction strength and population stability. Glob. Chang. Biol. 2010, 16, 2145–2157. [Google Scholar] [CrossRef]
- Berlow, E.L.; Dunne, J.A.; Martinez, N.D.; Stark, P.B.; Williams, R.J.; Brose, U. Simple prediction of interaction strengths in complex food webs. Proc. Natl. Acad. Sci. USA 2009, 106, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petchey, O.L.; Brose, U.; Rall, B.C. Predicting the effects of temperature on food web connectance. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2081–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culler, L.E.; Ayres, M.P.; Virginia, R.A. In a warmer Arctic, mosquitoes avoid increased mortality from predators by growing faster. Proc. R. Soc. B Biol. Sci. 2015, 282, 20151549. [Google Scholar] [CrossRef]
- Karban, R.; Grof-Tisza, P.; Mcmunn, M.; Kharouba, H.; Huntzinger, M. Caterpillars escape predation in habitat and thermal refuges. Ecol. Entomol. 2015, 40, 725–731. [Google Scholar] [CrossRef]
- Vucic-Pestic, O.; Ehnes, R.B.; Rall, B.C.; Brose, U. Warming up the system: Higher predator feeding rates but lower energetic efficiencies. Glob. Chang. Biol. 2011, 17, 1301–1310. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, H.; Xie, J.; Jiang, R.; Deng, C.; Pang, H. Transcriptome responses to heat-and cold-stress in ladybirds (Cryptolaemus montrouzieri Mulasnt) analyzed by deep-sequencing. Biol. Res. 2015, 48, 66. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.R.; Shan, Y.M.; Tan, Y.; Zhang, Z.R.; Pang, B.P. Comparative analysis of transcriptome responses to cold stress in Galeruca daurica (Coleoptera: Chrysomelidae). J. Insect Sci. 2019, 19, 8. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.A.L. Application of functional ‘Omics’ in environmental stress physiology: Insights, limitations, and future challenges. Curr. Opin. Insect Sci. 2014, 4, 35–41. [Google Scholar] [CrossRef]
- Dunning, L.T.; Dennis, A.B.; Sinclair, B.J.; Newcomb, R.D.; Buckley, T.R. Divergent transcriptional responses to low temperature among populations of alpine and lowland species of New Zealand stick insects (Micrarchus). Mol. Ecol. 2014, 23, 2712–2726. [Google Scholar] [CrossRef] [Green Version]
- Wei, D.; Jia, F.-X.; Tian, C.-B.; Tian, Y.; Smagghe, G.; Dou, W.; Wang, J.-J. Comparative proteomic analysis of Bactrocera dorsalis (Hendel) in response to thermal stress. J. Insect Physiol. 2015, 74, 16–24. [Google Scholar] [CrossRef]
- Liu, Y.; Su, H.; Li, R.; Li, X.; Xu, Y.; Dai, X.; Zhou, Y.; Wang, H. Comparative transcriptome analysis of Glyphodes pyloalis Walker (Lepidoptera: Pyralidae) reveals novel insights into heat stress tolerance in insects. BMC Genomics 2017, 18, 974. [Google Scholar] [CrossRef]
- Storey, K.B.; Storey, J.M. Insect cold hardiness: Metabolic, gene, and protein adaptation. This review is part of a virtual symposium on recent advances in understanding a variety of complex regulatory processes in insect physiology and endocrinology, including development, metaboli. Can. J. Zool. 2012, 90, 456–475. [Google Scholar] [CrossRef]
- Clark, M.S.; Worland, M.R. How insects survive the cold: Molecular mechanisms—A review. J. Comp. Physiol. B 2008, 178, 917–933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, D.; Zhang, Z.; He, X.; Liang, G. Validation of reference genes in Solenopsis invicta in different developmental stages, castes and tissues. PLoS ONE 2013, 8, e57718. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2004, 32, 115–119. [Google Scholar] [CrossRef]
- Powell, S.; Forslund, K.; Szklarczyk, D.; Trachana, K.; Roth, A.; Huerta-Cepas, J.; Gabaldón, T.; Rattei, T.; Creevey, C.; Kuhn, M.; et al. eggNOG v4.0: Nested orthology inference across 3686 organisms. Nucleic Acids Res. 2013, 42, 231–239. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Cui, M.; Hu, P.; Wang, T.; Tao, J.; Zong, S. Differential transcriptome analysis reveals genes related to cold tolerance in seabuckthorn carpenter moth, Eogystia hippophaecolus. PLoS ONE 2017, 12, e0187105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatanparast, M.; Park, Y. Comparison of gene expression in the red imported fire ant (Solenopsis invicta) under different temperature conditions. Sci. Rep. 2021, 11, 16476. [Google Scholar] [CrossRef] [PubMed]
- Ran, L.; Jiang, C.; Yang, Q.; Wang, H.; Bai ma, T.; Chen, L.; Kuang, J.; Huang, T.; Li, Q. Comparative transcriptome analysis of Locusta migratoria tibetensis Chen (Orthoptera: Oedipodidae) under high-and low-temperature stress. J. Appl. Entomol. 2020, 144, 181–190. [Google Scholar] [CrossRef]
- Tusong, K.; Lu, X.; Liu, X.; Ma, J. Transcriptomic analysis of the desert beetle Microdera punctipennis (Coleoptera: Tenebrionidae) in response to short-term cold stress. Acta Entomol. Sin. 2016, 59, 581–591. [Google Scholar]
- Robich, R.M.; Denlinger, D.L. Diapause in the mosquito Culex pipiens evokes a metabolic switch from blood feeding to sugar gluttony. Proc. Natl. Acad. Sci. USA 2005, 102, 15912–15917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanley-Samuelson, D.W.; Jurenka, R.A.; Cripps, C.; Blomquist, G.J.; de Renobales, M. Fatty acids in insects: Composition, metabolism, and biological significance. Arch. Insect Biochem. Physiol. 1988, 9, 1–33. [Google Scholar] [CrossRef]
- Da Costa, S.G.; Bates, P.; Dillon, R.; Genta, F.A. Characterization of α-glucosidases from Lutzomyia longipalpis reveals independent hydrolysis systems for plant or blood sugars. Front. Physiol. 2019, 10, 248. [Google Scholar] [CrossRef] [Green Version]
- MacMillan, H.A.; Knee, J.M.; Dennis, A.B.; Udaka, H.; Marshall, K.E.; Merritt, T.J.S.; Sinclair, B.J. Cold acclimation wholly reorganizes the Drosophila melanogaster transcriptome and metabolome. Sci. Rep. 2016, 6, 28999. [Google Scholar] [CrossRef] [Green Version]
- Overgaard, J.; Malmendal, A.; Sørensen, J.G.; Bundy, J.G.; Loeschcke, V.; Nielsen, N.C.; Holmstrup, M. Metabolomic profiling of rapid cold hardening and cold shock in Drosophila melanogaster. J. Insect Physiol. 2007, 53, 1218–1232. [Google Scholar] [CrossRef]
- Koštál, V.; Korbelová, J.; Rozsypal, J.; Zahradníčková, H.; Cimlová, J.; Tomčala, A.; Šimek, P. Long-term cold acclimation extends survival time at 0 °C and modifies the metabolomic profiles of the larvae of the fruit fly Drosophila melanogaster. PLoS ONE 2011, 6, e25025. [Google Scholar] [CrossRef] [Green Version]
- Colinet, H.; Larvor, V.; Laparie, M.; Renault, D. Exploring the plastic response to cold acclimation through metabolomics. Funct. Ecol. 2012, 26, 711–722. [Google Scholar] [CrossRef]
- Zhang, J.; Marshall, K.E.; Westwood, J.T.; Clark, M.S.; Sinclair, B.J. Divergent transcriptomic responses to repeated and single cold exposures in Drosophila melanogaster. J. Exp. Biol. 2011, 214, 4021–4029. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, C.J.; Sørensen, P.; Kirilova Gagalova, K.; Loeschcke, V. Transcriptomic analysis of inbreeding depression in cold-sensitive Drosophila melanogaster shows upregulation of the immune response. J. Evol. Biol. 2013, 26, 1890–1902. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; James, R.R. Temperature stress affects the expression of immune response genes in the alfalfa leafcutting bee, Megachile rotundata. Insect Mol. Biol. 2012, 21, 269–280. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, H.A.; Sinclair, B.J. The role of the gut in insect chilling injury: Cold-induced disruption of osmoregulation in the fall field cricket, Gryllus pennsylvanicus. J. Exp. Biol. 2011, 214, 726–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, S.; Alenius, M. Cis-regulatory mechanisms for robust olfactory sensory neuron class-restricted odorant receptor gene expression in Drosophila. PLoS Genet. 2015, 11, e1005051. [Google Scholar] [CrossRef]
- Nakashima, T.; Harada, Y.; Miyata, S.; Kiyohara, T. Inhibitors of cytochrome P-450 augment fever induced by interleukin-1 β. Am. J. Physiol. Integr. Comp. Physiol. 1996, 271, 1274–1279. [Google Scholar] [CrossRef]
- King, A.M.; MacRae, T.H. Insect heat shock proteins during stress and diapause. Annu. Rev. Entomol. 2014, 60, 59–75. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | Unigene | 300 ≤ Length < 1000 | Length ≥ 1000 |
|---|---|---|---|
| NT_Annotation | 29,598 (33.01%) | 11,806 | 11,029 |
| NR_Annotation | 32,782 (36.56%) | 12,648 | 10,156 |
| Pfam_Annotation | 20,267 (22.61%) | 6890 | 8242 |
| EggNOG_Annotation | 26,823 (29.92%) | 9520 | 9796 |
| KO_EUK_Annotation | 29,144 (32.51%) | 10,924 | 9738 |
| KO_PRO_Annotation | 6889 (7.68%) | 2126 | 3564 |
| KO_BAC_Annotation | 6376 (7.11%) | 1877 | 3469 |
| KO_BAC.NUC_Annotation | 242 (0.27%) | 79 | 115 |
| GO_Annotation | 20,645 (23.03%) | 6775 | 8254 |
| UniProt_Annotation | 19,121 (21.33%) | 6182 | 7782 |
| Assembly | Total Unigene | ORF Predicted Unigene | Single ORF Predicted Unigene | Multiple ORF Predicted Unigene | |
|---|---|---|---|---|---|
| Merge | 89,657 | 19,384 (21.62%) | 16,956 (87.47%) | 2428 (12.53%) | |
| Assembly | No. of ORF | Complete | Internal | 5′ partial | 3′ partial |
| Merge | 22,442 | 9904 (44.13%) | 6116 (27,25%) | 4790 (21.34%) | 1632 (7.27%) |
| GO Term | Unigenes | DEGs (FC ≥ 2, FC ≤ −2) | Corrected p-Value |
|---|---|---|---|
| Biological Process | |||
| Cellular process | 13,159 (24.32%) | 109 (22.52%) | 0.003627 |
| Metabolic process | 9319 (17.22%) | 110 (22.73%) | 4.8 × 10−5 |
| Biological regulation | 8026 (14.83%) | 52 (10.74%) | 0.004567 |
| Developmental process | 3838 (7.09%) | 23 (4.75%) | 0.002454 |
| Cellular component | 3971 (7.45%) | 65 (1.63%) | 2.85 × 10−5 |
| Localization | 3515 (6.5%) | 36 (7.44%) | 0.000132 |
| Cellular Component | |||
| Cell part | 14,694 (31.76%) | 132 (30.91%) | 0.000105 |
| Organelle | 7513 (16.24%) | 45 (10.54%) | 0.00182 |
| Membrane part | 4536 (9.8%) | 65 (15.22%) | 0.036674 |
| Protein-containing complex | 5210 (11.26%) | 26 (6.09%) | 0.000455 |
| Membrane | 3978 (8.6%) | 55 (12.88%) | 0.004567 |
| Organelle part | 6190 (13.38%) | 45 (10.54%) | 1.02 × 10−9 |
| Molecular Function | |||
| Binding | 10,355 (41.73%) | 97 (33.22%) | 3.29 × 10−14 |
| Catalytic activity | 8531 (34.38%) | 124 (42.47%) | 0.00136 |
| Transporter activity | 1565 (6.31%) | 36 (12.33%) | 5.71 × 10−6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vatanparast, M.; Park, Y. Comparative RNA-Seq Analyses of Solenopsis japonica (Hymenoptera: Formicidae) Reveal Gene in Response to Cold Stress. Genes 2021, 12, 1610. https://doi.org/10.3390/genes12101610
Vatanparast M, Park Y. Comparative RNA-Seq Analyses of Solenopsis japonica (Hymenoptera: Formicidae) Reveal Gene in Response to Cold Stress. Genes. 2021; 12(10):1610. https://doi.org/10.3390/genes12101610
Chicago/Turabian StyleVatanparast, Mohammad, and Youngjin Park. 2021. "Comparative RNA-Seq Analyses of Solenopsis japonica (Hymenoptera: Formicidae) Reveal Gene in Response to Cold Stress" Genes 12, no. 10: 1610. https://doi.org/10.3390/genes12101610
APA StyleVatanparast, M., & Park, Y. (2021). Comparative RNA-Seq Analyses of Solenopsis japonica (Hymenoptera: Formicidae) Reveal Gene in Response to Cold Stress. Genes, 12(10), 1610. https://doi.org/10.3390/genes12101610