
DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells


Abstract
:1. Introduction
2. Cell Cycle Regulation in Pluripotent Stem Cells (PSCs)
2.1. Cell Cycle Regulation in mESCs
2.2. Cell Cycle Regulation in hESCs
2.3. Cell Cycle Regulation and Pluripotency
3. DNA Damage Response and Cell Cycle Regulation
3.1. Cell Cycle Checkpoints and DNA Damage Response
3.2. DNA Repairing Mechanisms in PSCs
4. Critical Regulators of DNA Damage Response in PSCs
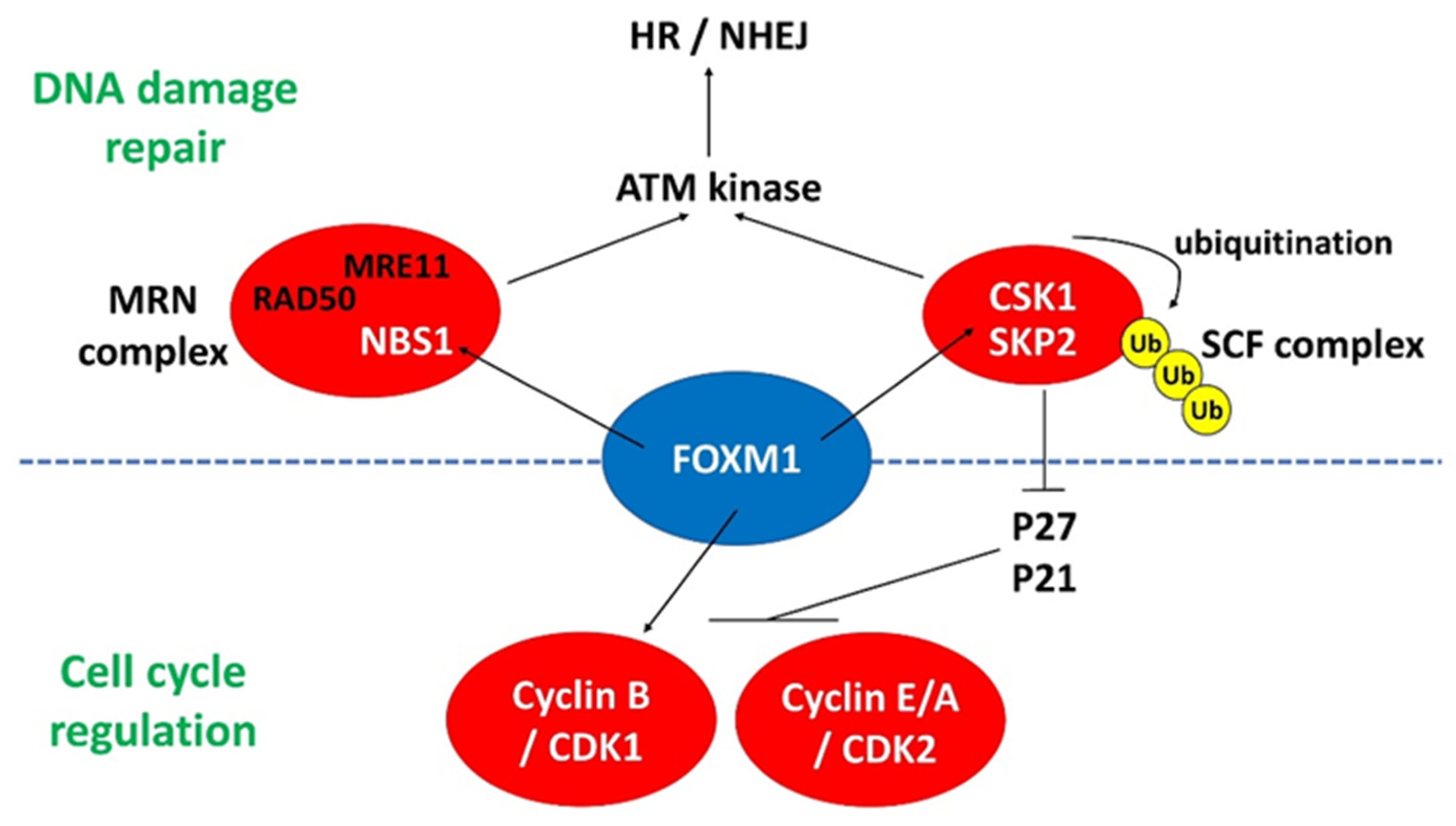
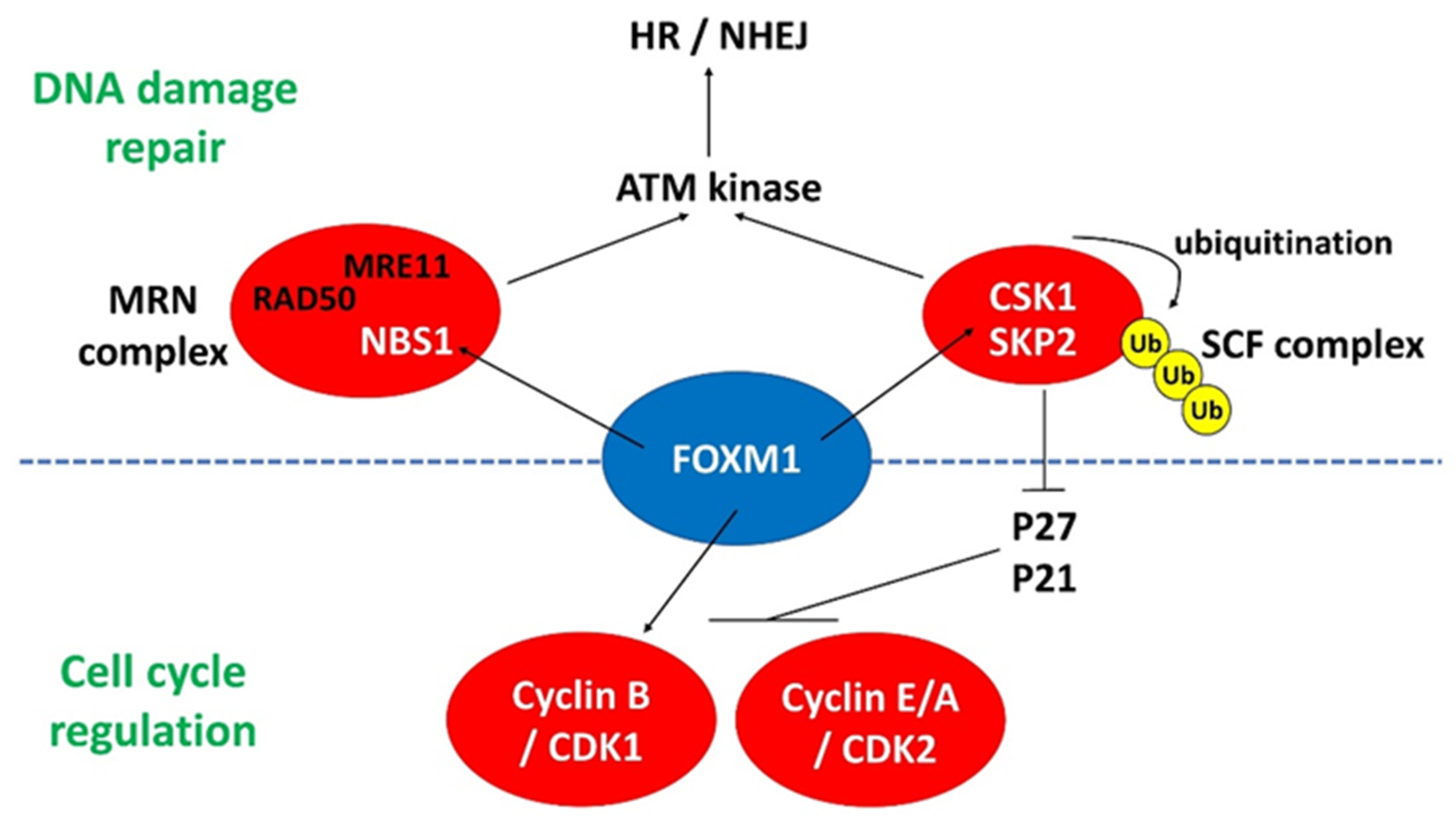
4.1. FOXM1
4.1.1. FOXM1 and DNA Damage Response in PSCs
4.1.2. FOXM1 and CDKs in PSCs
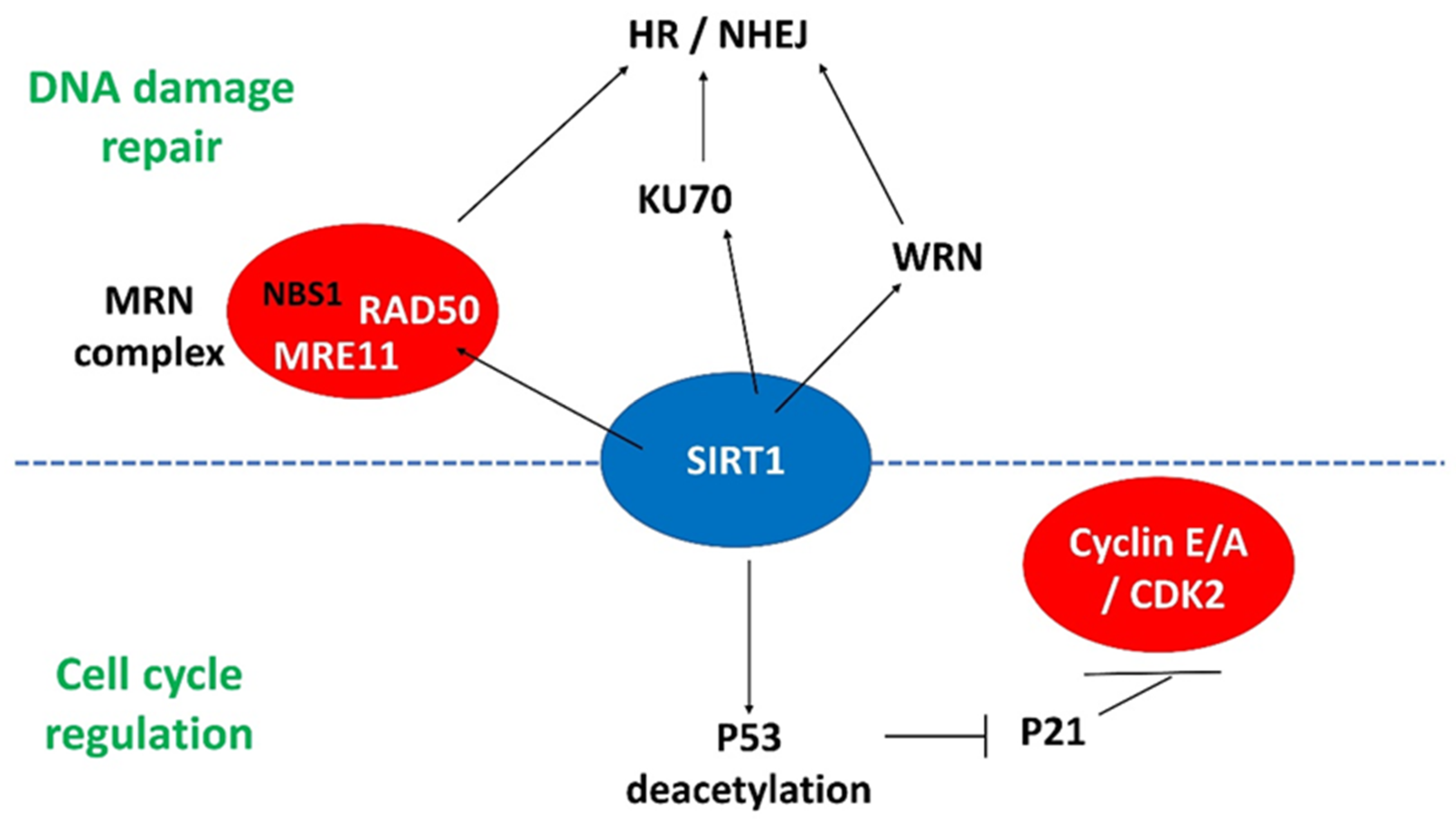
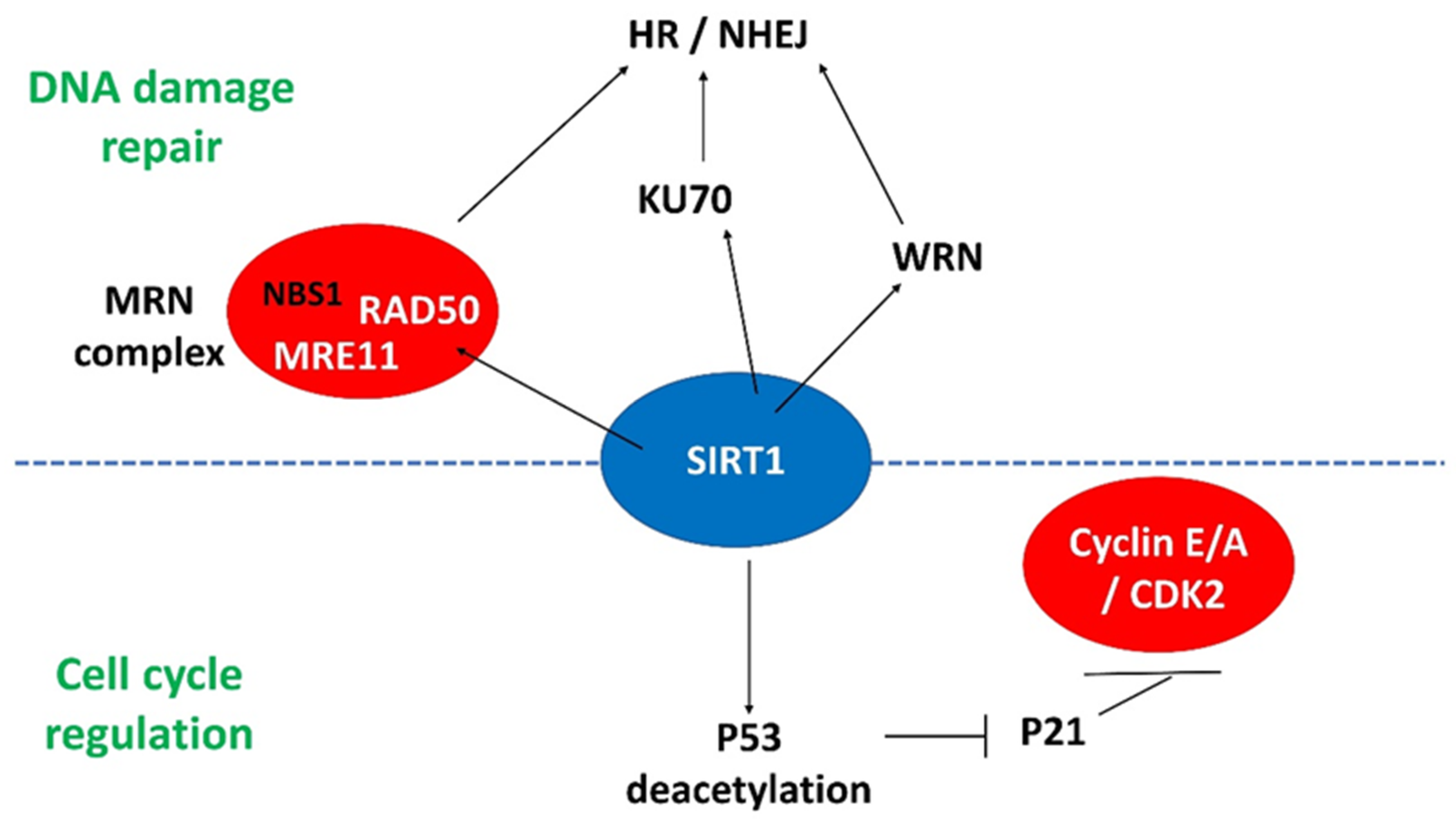
4.2. SIRT1
4.2.1. SIRT1 and P53
4.2.2. SIRT1 and DNA Damage Response Genes
4.3. Interplay between FOMX1 and SIRT1
4.4. PUMA
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, M.J.; Kaufman, M.H. Establishment in culture of pluripotential cells from mouse embryos. Nature 1981, 292, 154–156. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.C.H.; Lee, Y.L.; Fong, S.W.; Wong, C.C.Y.; Ng, E.; Yeung, S.B.W. Hyperglycemia impedes definitive endoderm differentiation of human embryonic stem cells by modulating histone methylation patterns. Cell Tissue Res. 2017, 368, 563–578. [Google Scholar] [CrossRef]
- Russ, A.H.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Rezania, A.; Bruin, J.E.; Riedel, M.J.; Mojibian, M.; Asadi, A.; Xu, J.; Gauvin, R.; Narayan, K.; Karanu, F.; O’Neil, J.J.; et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012, 61, 2016–2029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amita, M.; Adachi, K.; Alexenko, A.P.; Sinha, S.; Schust, D.J.; Schulz, L.; Roberts, R.M.; Ezashi, T. Complete and unidirectional conversion of human embryonic stem cells to trophoblast by BMP4. Proc. Natl. Acad. Sci. USA 2013, 110, E1212–E1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.L.; Fong, S.-W.; Chen, A.C.; Li, T.; Yue, C.; Lee, C.-L.; Ng, E.; Yeung, S.B.W.; Lee, K.-F. Establishment of a novel human embryonic stem cell-derived trophoblastic spheroid implantation model. Hum. Reprod. 2015, 30, 2614–2626. [Google Scholar] [CrossRef] [Green Version]
- Irie, N.; Weinberger, L.; Tang, W.W.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietmann, S.; Hanna, J.H.; Surani, M.A. SOX17 Is a Critical Specifier of Human Primordial Germ Cell Fate. Cell 2015, 160, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubi, J.A.; Chen, A.C.; Fong, S.W.; Lai, K.P.; Wong, C.K.; Yeung, W.S.; Lee, K.F.; Lee, Y.L. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on the differentiation of embryonic stem cells towards pancreatic lineage and pancreatic beta cell function. Environ. Int. 2019, 130, 104885. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.; Dalton, S. Cell Cycle Control of Embryonic Stem Cells. Stem Cell Rev. Rep. 2005, 1, 131–138. [Google Scholar] [CrossRef]
- Becker, K.A.; Ghule, P.N.; Therrien, J.A.; Lian, J.B.; Stein, J.L.; van Wijnen, A.J.; Stein, G.S. Self-renewal of human embryonic stem cells is supported by a shortened G1 cell cycle phase. J. Cell. Physiol. 2006, 209, 883–893. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Loewke, K.E.; Bossert, N.L.; Behr, B.; De Jonge, C.J.; Baer, T.M.; Reijo Pera, R.A. Non-invasive imaging of human embryos before embryonic genome activation predicts development to the blastocyst stage. Nat. Biotechnol. 2010, 28, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Neganova, I.; Zhang, X.; Atkinson, S.; Lako, M. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene 2009, 28, 20–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lange, C.; Calegari, F. Cdks and cyclins link G1length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle 2010, 9, 1893–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Go, Y.; Kang, I.; Han, Y.; Kim, J. Oct-4 controls cell-cycle progression of embryonic stem cells. Biochem. J. 2010, 426, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- Ahuja, A.K.; Jodkowska, K.; Teloni, F.; Bizard, A.H.; Zellweger, R.; Herrador, R.; Ortega, S.; Hickson, I.D.; Altmeyer, M.; Mendez, J.; et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 2016, 7, 10660. [Google Scholar] [CrossRef] [Green Version]
- Vallabhaneni, H.; Lynch, P.J.; Chen, G.; Park, K.; Liu, Y.; Goehe, R.; Mallon, B.S.; Boehm, M.; Hursh, D.A. High Basal Levels of γH2AX in Human Induced Pluripotent Stem Cells Are Linked to Replication-Associated DNA Damage and Repair. Stem. Cells 2018, 36, 1501–1513. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.-H.; Yoon, S.; Koh, Y.E.; Seo, Y.-J.; Kim, K.P. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp. Mol. Med. 2020, 52, 1220–1229. [Google Scholar] [CrossRef]
- Sherr, C.J. Cancer Cell Cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciemerych, M.A.; Sicinski, P. Cell cycle in mouse development. Oncogene 2005, 24, 2877–2898. [Google Scholar] [CrossRef] [Green Version]
- ter Huurne, M.; Chappell, J.; Dalton, S.; Stunnenberg, H.G. Distinct Cell-Cycle Control in Two Different States of Mouse Pluripotency. Cell Stem. Cell 2017, 21, 449–455.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii-Yamamoto, H.; Kim, J.M.; Arai, K.-I.; Masai, H. Cell Cycle and Developmental Regulations of Replication Factors in Mouse Embryonic Stem Cells. J. Biol. Chem. 2005, 280, 12976–12987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brehm, A.; Miska, E.; McCance, D.J.; Reid, J.L.; Bannister, A.; Kouzarides, T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nat. Cell Biol. 1998, 391, 597–601. [Google Scholar] [CrossRef]
- Stead, E.; White, J.; Faast, R.; Conn, S.; Goldstone, S.; Rathjen, J.; Dhingra, U.; Rathjen, P.; Walker, D.; Dalton, S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 2002, 21, 8320–8333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lai, N. Pluripotent States of Human Embryonic Stem Cells. Cell. Reprogramming 2015, 17, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, G.; Chang, Z.Y.; Schöler, H.; Pei, D. Stem cell pluripotency and transcription factor Oct4. Cell Res. 2002, 12, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeineddine, D.; Papadimou, E.; Chebli, K.; Gineste, M.; Liu, J.; Grey, C.; Thurig, S.; Behfar, A.; Wallace, V.A.; Skerjanc, I.S.; et al. Oct-3/4 Dose Dependently Regulates Specification of Embryonic Stem Cells toward a Cardiac Lineage and Early Heart Development. Dev. Cell 2006, 11, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Zafarana, G.; Avery, S.R.; Avery, K.; Moore, H.D.; Andrews, P.W. Specific Knockdown of OCT4 in Human Embryonic Stem Cells by Inducible Short Hairpin RNA Interference. Stem. Cells 2009, 27, 776–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Card, D.A.G.; Hebbar, P.B.; Li, L.; Trotter, K.W.; Komatsu, Y.; Mishina, Y.; Archer, T.K. Oct4/Sox2-Regulated miR-302 Targets Cyclin D1 in Human Embryonic Stem Cells. Mol. Cell. Biol. 2008, 28, 6426–6438. [Google Scholar] [CrossRef] [Green Version]
- Kanai, D.; Ueda, A.; Akagi, T.; Yokota, T.; Koide, H. Oct3/4 directly regulates expression of E2F3a in mouse embryonic stem cells. Biochem. Biophys. Res. Commun. 2015, 459, 374–378. [Google Scholar] [CrossRef]
- Julian, L.M.; Vandenbosch, R.; Pakenham, C.A.; Andrusiak, M.G.; Nguyen, A.P.; McClellan, K.A.; Svoboda, D.S.; Lagace, D.C.; Park, D.; Leone, G.; et al. Opposing Regulation of Sox2 by Cell-Cycle Effectors E2f3a and E2f3b in Neural Stem Cells. Cell Stem. Cell 2013, 12, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Van Der Laan, S.; Golfetto, E.; Vanacker, J.-M.; Maiorano, D. Cell Cycle-Dependent Expression of Dub3, Nanog and the p160 Family of Nuclear Receptor Coactivators (NCoAs) in Mouse Embryonic Stem Cells. PLoS ONE 2014, 9, e93663. [Google Scholar] [CrossRef] [PubMed]
- Faast, R.; White, J.; Cartwright, P.; Crocker, L.; Sarcevic, B.; Dalton, S. Cdk6–cyclin D3 activity in murine ES cells is resistant to inhibition by p16INK4a. Oncogene 2004, 23, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.A.; Xiong, J. p53 Amino-Terminal Nuclear Export Signal Inhibited by DNA Damage-Induced Phosphorylation. Science 2001, 292, 1910–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sancar, A.; Lindsey-Boltz, L.; Ünsal-Kaçmaz, K.; Linn, S. Molecular Mechanisms of Mammalian DNA Repair and the DNA Damage Checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, C.B.; Allen, F.; Crisp, A.; Grandy, R.A.; Vallier, L.; Sale, J.E. A p53-Dependent Checkpoint Induced upon DNA Damage Alters Cell Fate during hiPSC Differentiation. Stem. Cell Rep. 2020, 15, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Hyka-Nouspikel, N.; Desmarais, J.; Gokhale, P.J.; Jones, M.; Meuth, M.; Andrews, P.W.; Nouspikel, T. Deficient DNA Damage Response and Cell Cycle Checkpoints Lead to Accumulation of Point Mutations in Human Embryonic Stem Cells. Stem. Cells 2012, 30, 1901–1910. [Google Scholar] [CrossRef] [PubMed]
- Orford, K.W.; Scadden, D.T. Deconstructing stem cell self-renewal: Genetic insights into cell-cycle regulation. Nat. Rev. Genet. 2008, 9, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.W.-F.; Brosens, J.; Gomes, A.R.; Koo, C.Y. Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 2013, 13, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Kelly, T.F.; Samadani, U.; Lim, L.; Rubio, S.; Overdier, D.G.; A Roebuck, K.; Costa, R.H. Hepatocyte nuclear factor 3/fork head homolog 11 is expressed in proliferating epithelial and mesenchymal cells of embryonic and adult tissues. Mol. Cell. Biol. 1997, 17, 1626–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gemenetzidis, E.; Bose, A.; Riaz, A.M.; Chaplin, T.; Young, B.D.; Ali, M.; Sugden, D.; Thurlow, J.K.; Cheong, S.-C.; Teo, S.-H.; et al. FOXM1 Upregulation Is an Early Event in Human Squamous Cell Carcinoma and it Is Enhanced by Nicotine during Malignant Transformation. PLoS ONE 2009, 4, e4849. [Google Scholar] [CrossRef] [PubMed]
- Bella, L.; Zona, S.; de Moraes, G.N.; Lam, E.W.-F. FOXM1: A key oncofoetal transcription factor in health and disease. Semin. Cancer Biol. 2014, 29, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.D.; Leung, M.H.; Qin, J.; Qin, Y.; Wang, J.; Lee, Y.L.; Yao, K.-M. The Forkhead box transcription factor FOXM1 is required for the maintenance of cell proliferation and protection against oxidative stress in human embryonic stem cells. Stem. Cell Res. 2016, 16, 651–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Raychaudhuri, P.; Costa, R.H. Chk2 Mediates Stabilization of the FoxM1 Transcription Factor To Stimulate Expression of DNA Repair Genes. Mol. Cell. Biol. 2007, 27, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Khongkow, P.; Karunarathna, U.; Gong, C.; Gomes, A.R.; Yagüe, E.; Monteiro, L.J.; Kongsema, M.; Zona, S.; Man, E.P.S.; Tsang, J.W.-H.; et al. FOXM1 targets NBS1 to regulate DNA damage-induced senescence and epirubicin resistance. Oncogene 2014, 33, 4144–4155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, I.-C.; Chen, Y.-J.; Hughes, D.; Petrovic, V.; Major, M.L.; Park, H.J.; Tan, Y.; Ackerson, T.; Costa, R.H. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol. Cell. Biol. 2005, 25, 10875–10894. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhang, X.; Zhang, L.; Wu, C.-Y.; Rezaeian, A.H.; Chan, C.-H.; Li, J.-M.; Wang, J.; Gao, Y.; Han, F.; et al. Skp2 E3 Ligase Integrates ATM Activation and Homologous Recombination Repair by Ubiquitinating NBS1. Mol. Cell 2012, 46, 351–361. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, P.; Dodsworth, B.T.; Sidders, B.; Gutteridge, A.; Michaelides, C.; Duckworth, J.K.; Whiting, P.; Benn, C.L. A FOXM1 Dependent Mesenchymal-Epithelial Transition in Retinal Pigment Epithelium Cells. PLoS ONE 2015, 10, e0130379. [Google Scholar] [CrossRef] [PubMed]
- Vassilopoulos, A.; Fritz, K.S.; Petersen, D.R.; Gius, D. The human sirtuin family: Evolutionary divergences and functions. Hum. Genom. 2011, 5, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, A.C.H.; Peng, Q.; Fong, S.W.; Yeung, W.S.B.; Lee, Y.L. Sirt1 is regulated by miR-135a and involved in DNA damage repair during mouse cellular reprogramming. Aging 2020, 12, 7431–7447. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.L.; Peng, Q.; Fong, S.W.; Chen, A.C.H.; Lee, K.F.; Ng, E.; Nagy, A.; Yeung, W.S.B. Sirtuin 1 Facilitates Generation of Induced Pluripotent Stem Cells from Mouse Embryonic Fibroblasts through the miR-34a and p53 Pathways. PLoS ONE 2012, 7, e45633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-C.; Guarente, L. SIRT1 Mediates Central Circadian Control in the SCN by a Mechanism that Decays with Aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.; Yamada, K.A.; Imai, S.-I. Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Vaquero, A.; Scher, M.; Lee, D.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Human SirT1 Interacts with Histone H1 and Promotes Formation of Facultative Heterochromatin. Mol. Cell 2004, 16, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Guarente, L.; Picard, F. Calorie Restriction—the SIR2 Connection. Cell 2005, 120, 473–482. [Google Scholar] [CrossRef] [Green Version]
- Calvanese, V.; Lara, E.; Suárez-Álvarez, B.; Abu Dawud, R.; Vázquez-Chantada, M.; Martínez-Chantar, M.L.; Embade, N.; López-Nieva, P.; Horrillo, A.; Hmadcha, A.; et al. Sirtuin 1 regulation of developmental genes during differentiation of stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13736–13741. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-N.; Chung, S.-K.; Xu, Z.; Xu, Y. Oct4 Maintains the Pluripotency of Human Embryonic Stem Cells by Inactivating p53 Through Sirt1-Mediated Deacetylation. Stem. Cells 2014, 32, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.; Huh, Y.J.; Cho, H.-J.; Lee, B.R.; Park, J.; Hwang, D.-Y.; Kim, D.-W. SIRT1 Enhances the Survival of Human Embryonic Stem Cells by Promoting DNA Repair. Stem. Cell Rep. 2017, 9, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Zhang, X.; Sengupta, N.; Lane, W.S.; Seto, E. SIRT1 Regulates the Function of the Nijmegen Breakage Syndrome Protein. Mol. Cell 2007, 27, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Casta, A.; Wang, R.; Lozada, E.; Fan, W.; Kane, S.; Ge, Q.; Gu, W.; Orren, D.; Luo, J. Regulation of WRN Protein Cellular Localization and Enzymatic Activities by SIRT1-mediated Deacetylation. J. Biol. Chem. 2008, 283, 7590–7598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-Y.; Lee, H.; Kim, E.-S.; Park, S.; Lee, J.; Ahn, B. WRN translocation from nucleolus to nucleoplasm is regulated by SIRT1 and required for DNA repair and the development of chemoresistance. Mutat. Res. Mol. Mech. Mutagen. 2015, 774, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.L.; Ghosh, A.K.; Bohr, V.A. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair 2010, 9, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Chen, J. MRE11-RAD50-NBS1 Complex Dictates DNA Repair Independent of H2AX. J. Biol. Chem. 2010, 285, 1097–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-J.; Lee, H.J.; Son, B.-H.; Kim, S.-B.; Ahn, J.-H.; Ahn, S.D.; Cho, E.Y.; Gong, G. Expression of FOXM1 and related proteins in breast cancer molecular subtypes. Int. J. Exp. Pathol. 2016, 97, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, C.M.; Lu, T.Y.; Bacigalupa, Z.A.; Katsetos, C.D.; Sinclair, D.; Reginato, M.J. O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene 2017, 36, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, C.; Zhao, G.; Sun, X.; Wang, P.; Xie, N.; Luo, J.; Tong, T. Acetylation of FOXM1 is essential for its transactivation and tumor growth stimulation. Oncotarget 2016, 7, 60366–60382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, G.-Y.; Shi, B.-Z.; Li, Y. FoxM1 regulates Sirt1 expression in glioma cells. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 205–211. [Google Scholar] [PubMed]
- Qiu, W.; Carson-Walter, E.B.; Liu, H.; Epperly, M.; Greenberger, J.S.; Zambetti, G.P.; Zhang, L.; Yu, J. PUMA Regulates Intestinal Progenitor Cell Radiosensitivity and Gastrointestinal Syndrome. Cell Stem. Cell 2008, 2, 576–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Shen, H.; Yuan, Y.; XuFeng, R.; Hu, X.; Garrison, S.P.; Zhang, L.; Yu, J.; Zambetti, G.P.; Cheng, T. Deletion of Puma protects hematopoietic stem cells and confers long-term survival in response to high-dose γ-irradiation. Blood 2010, 115, 3472–3480. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Zhan, Z.; Ji, G.; Sang, Y.; Zhou, D.; Li, Y.; Feng, H.; Cheng, T. PUMA facilitates EMI1-promoted cytoplasmic Rad51 ubiquitination and inhibits DNA repair in stem and progenitor cells. Signal Transduct. Target. Ther. 2021, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; West, S.C. Role of the human RAD51 protein in homologous recombination and double-stranded-break repair. Trends Biochem. Sci. 1998, 23, 247–251. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| mESCs | hESCs | |
|---|---|---|
| Cyclin expression levels | ||
| Cyclin A/E | High, non-oscillatory | High, oscillatory |
| Cyclin B | High, oscillatory | High, oscillatory |
| Cyclin D | Low, oscillatory | Intermediate, oscillatory |
| CDKs expression levels | ||
| CDK1 | High, oscillatory | High, oscillatory |
| CDK2 | High, non-oscillatory | High, oscillatory |
| CDK4 | Low, oscillatory | Medium, oscillatory |
| CDK6 | Medium, oscillatory | Low, oscillatory |
| RB phosphorylation | ||
| RB | Hyper-phosphorylated | Hypo-/Hyper-phosphorylated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, A.C.H.; Peng, Q.; Fong, S.W.; Lee, K.C.; Yeung, W.S.B.; Lee, Y.L. DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes 2021, 12, 1548. https://doi.org/10.3390/genes12101548
Chen ACH, Peng Q, Fong SW, Lee KC, Yeung WSB, Lee YL. DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes. 2021; 12(10):1548. https://doi.org/10.3390/genes12101548
Chicago/Turabian StyleChen, Andy Chun Hang, Qian Peng, Sze Wan Fong, Kai Chuen Lee, William Shu Biu Yeung, and Yin Lau Lee. 2021. "DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells" Genes 12, no. 10: 1548. https://doi.org/10.3390/genes12101548
APA StyleChen, A. C. H., Peng, Q., Fong, S. W., Lee, K. C., Yeung, W. S. B., & Lee, Y. L. (2021). DNA Damage Response and Cell Cycle Regulation in Pluripotent Stem Cells. Genes, 12(10), 1548. https://doi.org/10.3390/genes12101548