
Global Transcriptome Characterization and Assembly of the Thermophilic Ascomycete Chaetomium thermophilum


Abstract
:1. Introduction
2. Materials and Methods
2.1. RNA Isolation and Sequencing
2.2. RNA Sequencing Data Analysis
2.3. Sequence Conservation and GO Annotation
2.4. Isoform Annotation
3. Results
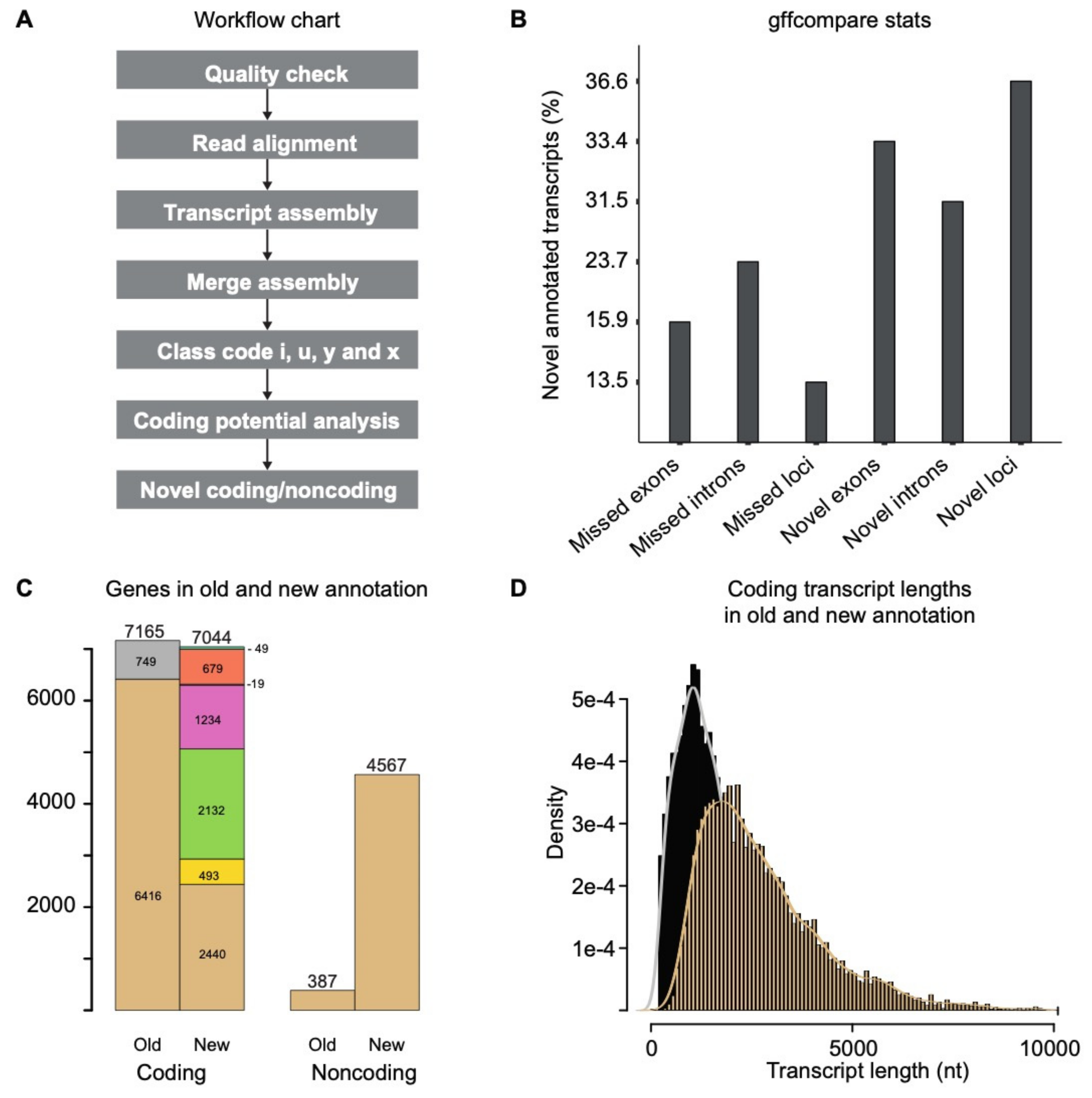
3.1. Transcripts Reassembly and Identification of Novel Transcripts
3.2. Annotation of the Novel Noncoding RNAs in C. Thermophilum
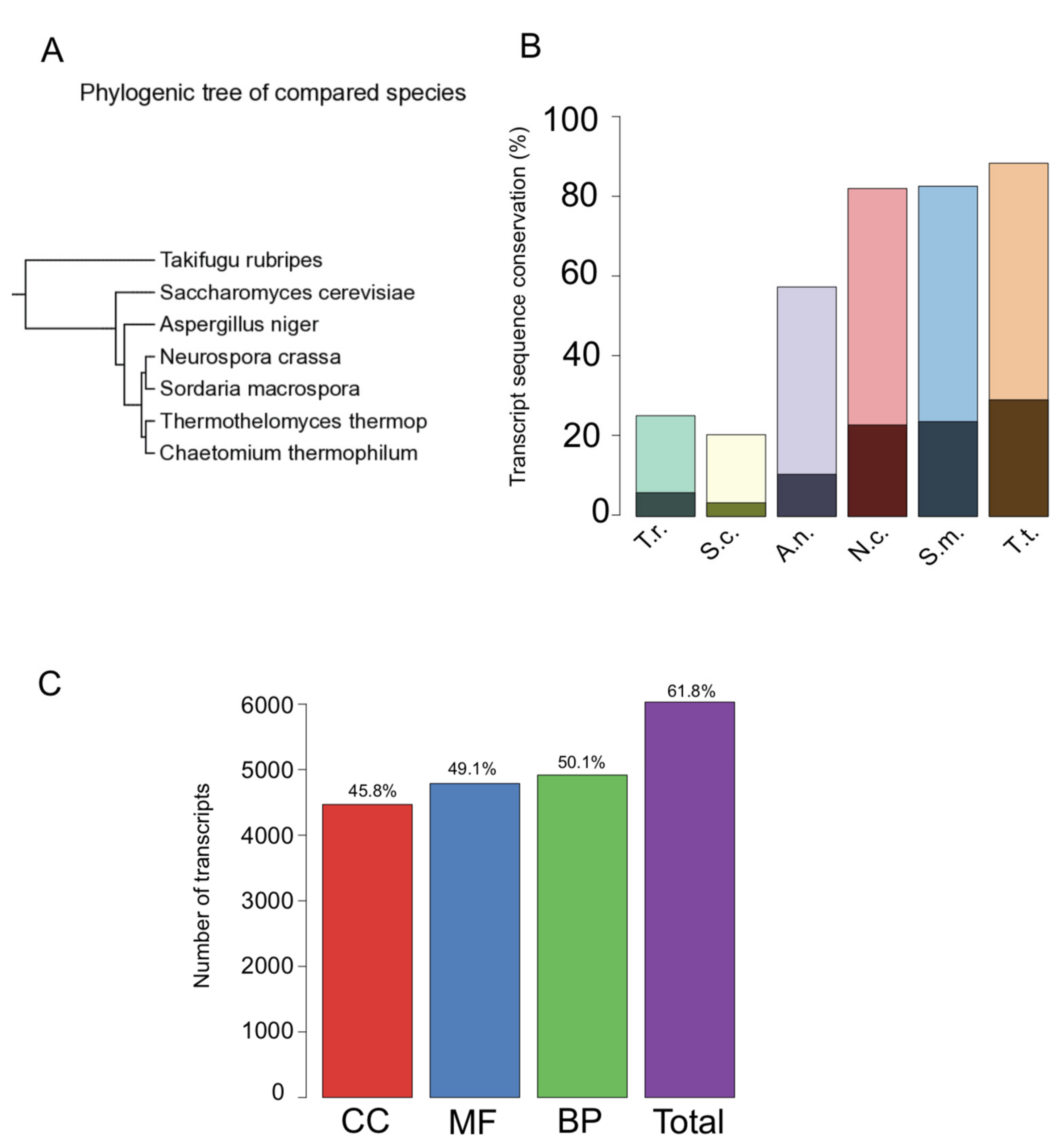
3.3. Sequence Conservation and Functional Annotation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Accession Numbers
Appendix A

{kind=link}
{kind=link}
{kind=link}
| (A) | ||
| Sample Name | Input Read | Overall Alignment Rate |
| G1 | 69350606 | 95.94% |
| G2 | 73230226 | 95.87% |
| G3 | 59976502 | 95.83% |
| (B) | ||
| Class Code | Description | Total Annotation |
| = | Complete match of intron chain | 3078 |
| c | Contained in reference (and intron isoform compatible) | 323 |
| k | Containment of reference (reverse containment) | 0 |
| j | At least one splice junction match | 4234 |
| e | At single exon, overlapping intron a possibly pre-mRNA fragment (un spliced intron) | 420 |
| o | Other same strand overlap with reference exons | 1896 |
| s | Intron match on the opposite strand (likely a mapping error) | 158 |
| x | Exonic overlap on the opposite strand (like ‘o’ or ‘e’, but on the opposite strand) | 1754 |
| i | Fully contained in a reference intron | 28 |
| y | Contains a reference within is intron(s) | 5 |
| p | Possible polymerase run-on (no actual overlap) | 706 |
| r | Repeat (at least 50% bases soft masked) | 0 |
| u | None of the above (unknown, intergenic) | 2744 |
| Software | Usage | Parameter Settings/Webpage |
|---|---|---|
| Ensemble genome (version 2.8) | C. thermophilum reference genome used for the analysis | https://fungi.ensembl.org, accessed on 17 August 2021 |
| FastQC (Version: 0.11.5) | The data quality assessment of raw sequence data | http://www.bioinformatics. babraham.ac.uk/projects/fastqc/, accessed on 17 August 2021 |
| HISAT2 (Version: 1.3.3b) | The raw reads were mapped to C. thermophilum reference genome | [hisat2 -p 8 -x -max-intronlen 2000 -dta-U]) |
| Stringtie (v1.3.4 release) | The mapped reads from HISAT2 for each sample were assembled separately | [stringtie -o -m 50 -p 8 -j 3 -c 5 -g 15]) |
| GffCompare (Version: v0.10.1) | The program used to compare, merge, annotate, and estimate accuracy “query” files, when compared with a reference annotation | gffcompare-merge -K -o gffcomp -i |
| CPC2 | The CPC2 calculate the coding or noncoding of the transcript | CPC2.py -i. Input.fasta -o output.txt |
| dc-mega BLAST (Version: 2.7.1+) | Sequence conservation analysis between different species | https://blast.ncbi.nlm.nih.gov, accessed on 17 August 2021 |
| Blast2GO (Version 5.1.1) | Functional annotation such as GO term and EC number are extracted from the software | http://docs.blast2go.com/, accessed on 17 August 2021 |
| blastx. | Finds regions of local similarity between sequences | https://blast.ncbi.nlm.nih.gov/, accessed on 17 August 2021 |
| T-Coffee software | Pairwise similarity score are calculated | http://tcoffee.crg.cat/, accessed on 17 August 2021 |
| phyloT | Visualization of phylogenetic tree different species | https://phylot.biobyte.de, accessed on 17 August 2021 |
| pyfaidx python package | Allowing for fast random access to any subsequence in the indexed FASTA file | https://pypi.org/project/pyfaidx/, accessed on 17 August 2021 |
| R (Version 3.3.3) | Creating figures and GO annotation package | http://www.R-project.org/, accessed on 17 August 2021 |
References
- La Touche, G. A Chaetomium-like thermophile fungus. Nature 1948, 161, 320. [Google Scholar] [CrossRef]
- Ganju, R.K.; Vithayathil, P.J.; Murthy, S. Purification and characterization of two xylanases from Chaetomium thermophile var. Coprophile. Can. J. Microbiol. 1989, 35, 836–842. [Google Scholar] [CrossRef]
- Chefetz, B.; Chen, Y.; Hadar, Y. Purification and characterization of laccase from Chaetomium thermophilium and its role in humification. Appl. Environ. Microbiol. 1998, 64, 3175–3179. [Google Scholar] [CrossRef] [Green Version]
- Li, D.-C.; Lu, M.; Li, Y.-L.; Lu, J. Purification and characterization of an endocellulase from the thermophilic fungus Chaetomium thermophilum CT. Enzym. Microb. Technol. 2003, 33, 932–937. [Google Scholar] [CrossRef]
- Li, A.-N.; Ding, A.-Y.; Chen, J.; Liu, S.-A.; Zhang, M.; Li, D.-C. Purification and characterization of two thermostable proteases from the thermophilic fungus Chaetomium thermophilum. J. Microbiol. Biotechnol. 2007, 17, 624–631. [Google Scholar] [PubMed]
- Li, A.-N.; Yu, K.; Liu, H.-Q.; Zhang, J.; Li, H.; Li, D. Two novel thermostable chitinase genes from thermophilic fungi: Cloning, expression and characterization. Bioresour. Technol. 2010, 101, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Hakulinen, N.; Turunen, O.; Jänis, J.; Leisola, M.; Rouvinen, J. Three-dimensional structures of thermophilic beta-1,4-xylanases from Chaetomium thermophilum and Nonomuraea flexuosa. Comparison of twelve xylanases in relation to their thermal stability. JBIC J. Biol. Inorg. Chem. 2003, 270, 1399–1412. [Google Scholar] [CrossRef] [Green Version]
- Rosgaard, L.; Pedersen, S.; Cherry, J.R.; Harris, P.; Meyer, A.S. Efficiency of New Fungal Cellulase Systems in Boosting Enzymatic Degradation of Barley Straw Lignocellulose. Biotechnol. Prog. 2006, 22, 493–498. [Google Scholar] [CrossRef]
- Voutilainen, S.P.; Puranen, T.; Siika-Aho, M.; Lappalainen, A.; Alapuranen, M.; Kallio, J.; Hooman, S.; Viikari, L.; Vehmaanperä, J.; Koivula, A. Cloning, expression, and characterization of novel thermostable family 7 cellobiohydrolases. Biotechnol. Bioeng. 2008, 101, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Elleuche, S.; Schäfers, C.; Blank, S.; Schröder, C.; Antranikian, G. Exploration of extremophiles for high temperature biotechnological processes. Curr. Opin. Microbiol. 2015, 25, 113–119. [Google Scholar] [CrossRef]
- Sriyapai, T.; Somyoonsap, P.; Matsui, K.; Kawai, F.; Chansiri, K. Cloning of a thermostable xylanase from Actinomadura sp. S14 and its expression in escherichia coli and pichia pastoris. J. Biosci. Bioeng. 2011, 111, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ji, P.; Zhao, Y.; Hua, C.; Han, C. Engineering the conserved and noncatalytic residues of a thermostable β-1, 4-endoglucanase to improve specific activity and thermostability. Sci. Rep. 2018, 8, 2954. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Ji, P.; Zhang, J.; Li, X.; Han, C. Characterization of a novel thermostable GH45 endoglucanase from Chaetomium thermophilum and its biodegradation of pectin. J. Biosci. Bioeng. 2017, 124, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ji, P.; Zhou, Q.; Hua, C.; Han, C. Insights into the Synergistic Biodegradation of Waste Papers Using a Combination of Thermostable Endoglucanase and Cellobiohydrolase from Chaetomium thermophilum. Mol. Biotechnol. 2017, 60, 49–54. [Google Scholar] [CrossRef]
- Monecke, T.; Haselbach, D.; Voß, B.; Russek, A.; Neumann, P.; Thomson, E.; Hurt, E.; Zachariae, U.; Stark, H.; Grubmüller, H.; et al. Structural basis for cooperativity of CRM1 export complex formation. Proc. Natl. Acad. Sci. USA 2012, 110, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Ulrich, A.; Wahl, M.C. Structure and evolution of the spliceosomal peptidyl-prolyl cis–trans isomerase cwc27. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 3110–3123. [Google Scholar] [CrossRef]
- Aibara, S.; Valkov, E.; Lamers, M.H.; Dimitrova, L.; Hurt, E.; Stewart, M. Structural characterization of the principal mRNA-export factor mex67–mtr2 from Chaetomium thermophilum. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 876–888. [Google Scholar] [CrossRef]
- Hondele, M.; Stuwe, T.; Hassler, M.; Halbach, F.; Bowman, A.; Zhang, E.T.; Nijmeijer, B.; Kotthoff, C.; Rybin, V.; Amlacher, S.; et al. Structural basis of histone H2A–H2B recognition by the essential chaperone FACT. Nature 2013, 499, 111–114. [Google Scholar] [CrossRef]
- Leidig, C.; Bange, G.; Kopp, J.; Amlacher, S.; Aravind, A.; Wickles, S.; Witte, G.; Hurt, E.; Beckmann, R.; Sinning, I. Structural characterization of a eukaryotic chaperone—The ribosome-associated complex. Nat. Struct. Mol. Biol. 2012, 20, 23–28. [Google Scholar] [CrossRef]
- Baker, R.W.; Jeffrey, P.D.; Zick, M.; Phillips, B.P.; Wickner, W.T.; Hughson, F.M. A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science 2015, 349, 1111–1114. [Google Scholar] [CrossRef] [Green Version]
- Stuwe, T.; Bley, C.J.; Thierbach, K.; Petrovic, S.; Schilbach, S.; Mayo, D.J.; Perriches, T.; Rundlet, E.J.; Jeon, Y.E.; Collins, L.N.; et al. Architecture of the fungal nuclear pore inner ring complex. Science 2015, 350, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornprobst, M.; Turk, M.; Kellner, N.; Cheng, J.; Flemming, D.; Koš-Braun, I.; Kos, M.; Thoms, M.; Berninghausen, O.; Beckmann, R.; et al. Architecture of the 90S Pre-ribosome: A Structural View on the Birth of the Eukaryotic Ribosome. Cell 2016, 166, 380–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amlacher, S.; Sarges, P.; Flemming, D.; van Noort, V.; Kunze, R.; Devos, D.; Arumugam, M.; Bork, P.; Hurt, E. Insight into Structure and Assembly of the Nuclear Pore Complex by Utilizing the Genome of a Eukaryotic Thermophile. Cell 2011, 146, 277–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, T.K.C.; Jochen, B.; Ori, A.; Malik, N.; Silva-Martin, N.; Huerta-Cepas, J.; Powell, S.; Kastritis, P.; Smyshlyaev, G.; Vonkova, I.; et al. An integrated approach for genome annotation of the eukaryotic thermophile Chaetomium thermophilum. Nucleic Acids Res. 2014, 42, 13525–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; Van Baren, M.J.; Salzberg, S.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.; Mwenifumbo, J.C.; Goya, R.; Morrissy, A.S.; Morin, R.D.; Corbett, R.; Tang, M.J.; Hou, Y.-C.; Pugh, T.; et al. Alternative expression analysis by RNA sequencing. Nat. Methods 2010, 7, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, M.; Kück, U. Polymorphic karyotypes in related Acremonium strains. Curr. Genet. 1991, 19, 73–76. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-J.; Yang, D.-C.; Kong, L.; Hou, M.; Meng, Y.-Q.; Wei, L.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; Garcia-Gomez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014. [Google Scholar]
- Shirley, M.D.; Ma, Z.; Pedersen, B.S.; Wheelan, S.J. Efficient “pythonic” access to fasta files using pyfaidx. PeerJ PrePrints 2015, 3, e970v1. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, M.; Pages, H. AnnotationForge: Code for Building Annotation Database Packages, R package version 1.4.4.; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- Mäntylä, A.; Paloheimo, M.; Hakola, S.; Lindberg, E.; Leskinen, S.; Kallio, J.; Vehmaanperä, J.; Lantto, R.; Suominen, P. Production in trichoderma reesei of three xylanases from Chaetomium thermophilum: A recombinant thermoxylanase for biobleaching of kraft pulp. Appl. Microbiol. Biotechnol. 2007, 76, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-N.; Li, D.-C. Cloning, expression and characterization of the serine protease gene from Chaetomium thermophilum. J. Appl. Microbiol. 2009, 106, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Garre, V.; Müller, U.; Tudzynski, P. Cloning, Characterization, and Targeted Disruption of cpcat1, Coding for an in Planta Secreted Catalase of Claviceps purpurea. Mol. Plant-Microbe Interact. 1998, 11, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Zámocký, M.; Tafer, H.; Chovanová, K.; Lopandic, K.; Kamlárová, A.; Obinger, C. Genome sequence of the filamentous soil fungus Chaetomium cochliodes reveals abundance of genes for heme enzymes from all peroxidase and catalase superfamilies. BMC Genom. 2016, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-F.; Lehmann, L.; Lin, J.J.; Vashisht, A.; Schmidt, R.; Ferrari, R.; Huang, C.; McKee, R.; Mosley, A.; Plath, K.; et al. Mediator and SAGA Have Distinct Roles in Pol II Preinitiation Complex Assembly and Function. Cell Rep. 2012, 2, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Baßler, J.; Ahmed, Y.L.; Kallas, M.; Kornprobst, M.; Calviño, F.R.; Gnädig, M.; Thoms, M.; Stier, G.; Ismail, S.; Kharde, S.; et al. Interaction network of the ribosome assembly machinery from a eukaryotic thermophile. Protein Sci. 2017, 26, 327–342. [Google Scholar] [CrossRef] [Green Version]
- Brighenti, E.; Trere, D.; Derenzini, M. Targeted cancer therapy with ribosome biogenesis inhibitors: A real possibility. Oncotarget 2015, 6, 38617. [Google Scholar] [CrossRef] [Green Version]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2014, 1842, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Muehl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Hoelzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [Green Version]
- Consortium, U. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar]
- Finn, R.D.; Attwood, T.K.; Babbitt, P.C.; Bateman, A.; Bork, P.; Bridge, A.J.; Chang, H.-Y.; Dosztányi, Z.; El-Gebali, S.; Fraser, M.; et al. InterPro in 2017 Beyond protein family and domain annotations. Nucleic Acids Res. 2016, 45, D190–D199. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Mistry, J.; Schuster-Böckler, B.; Griffiths-Jones, S.; Hollich, V.; Lassmann, T.; Moxon, S.; Marshall, M.; Khanna, A.; Durbin, R.; et al. Pfam: Clans, web tools and services. Nucleic Acids Res. 2006, 34, D247–D251. [Google Scholar] [CrossRef] [Green Version]
- Coordinators, N.R. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2016, 44, D7. [Google Scholar]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The seed and the rapid annotation of microbial genomes using subsystems technology (rast). Nucleic Acids Res. 2013, 42, D206–D214. [Google Scholar] [CrossRef]
- Kamburov, A.; Wierling, C.; Lehrach, H.; Herwig, R. ConsensusPathDB—A database for integrating human functional interaction networks. Nucleic Acids Res. 2008, 37, D623–D628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi-Tope, G.; Gillespie, M.; Vastrik, I.; D’Eustachio, P.; Schmidt, E.; Bono, B.; de Jassal, B.; Gopinath, G.; Wu, G.; Matthews, L.; et al. Reactome: A knowledgebase of biological pathways. Nucleic Acids Res. 2005, 33, D428–D432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.K.; Bencurova, E.; Srivastava, M.; Pahlavan, P.; Balkenhol, J.; Dandekar, T. Improving re-annotation of annotated eukaryotic genomes. In Big Data Analytics in Genomics; Springer: Cham, Switzerland, 2016; pp. 171–195. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.; Schermann, G.; Reislöhner, S.; Kellner, N.; Hurt, E.; Brunner, M. Global Transcriptome Characterization and Assembly of the Thermophilic Ascomycete Chaetomium thermophilum. Genes 2021, 12, 1549. https://doi.org/10.3390/genes12101549
Singh A, Schermann G, Reislöhner S, Kellner N, Hurt E, Brunner M. Global Transcriptome Characterization and Assembly of the Thermophilic Ascomycete Chaetomium thermophilum. Genes. 2021; 12(10):1549. https://doi.org/10.3390/genes12101549
Chicago/Turabian StyleSingh, Amit, Géza Schermann, Sven Reislöhner, Nikola Kellner, Ed Hurt, and Michael Brunner. 2021. "Global Transcriptome Characterization and Assembly of the Thermophilic Ascomycete Chaetomium thermophilum" Genes 12, no. 10: 1549. https://doi.org/10.3390/genes12101549
APA StyleSingh, A., Schermann, G., Reislöhner, S., Kellner, N., Hurt, E., & Brunner, M. (2021). Global Transcriptome Characterization and Assembly of the Thermophilic Ascomycete Chaetomium thermophilum. Genes, 12(10), 1549. https://doi.org/10.3390/genes12101549