Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies


Abstract
1. Introduction
2. Materials and Methods
2.1. LGMD Patients and Clinical Examination
2.2. MicroRNAs Analysis
2.3. Muscle MRI
2.4. Statistical Analysis
3. Results
3.1. LGMD Clinical and Genetic Features
3.1.1. Transportinopathy
3.1.2. Calpainopathy
3.1.3. Sarcoglycanopathy
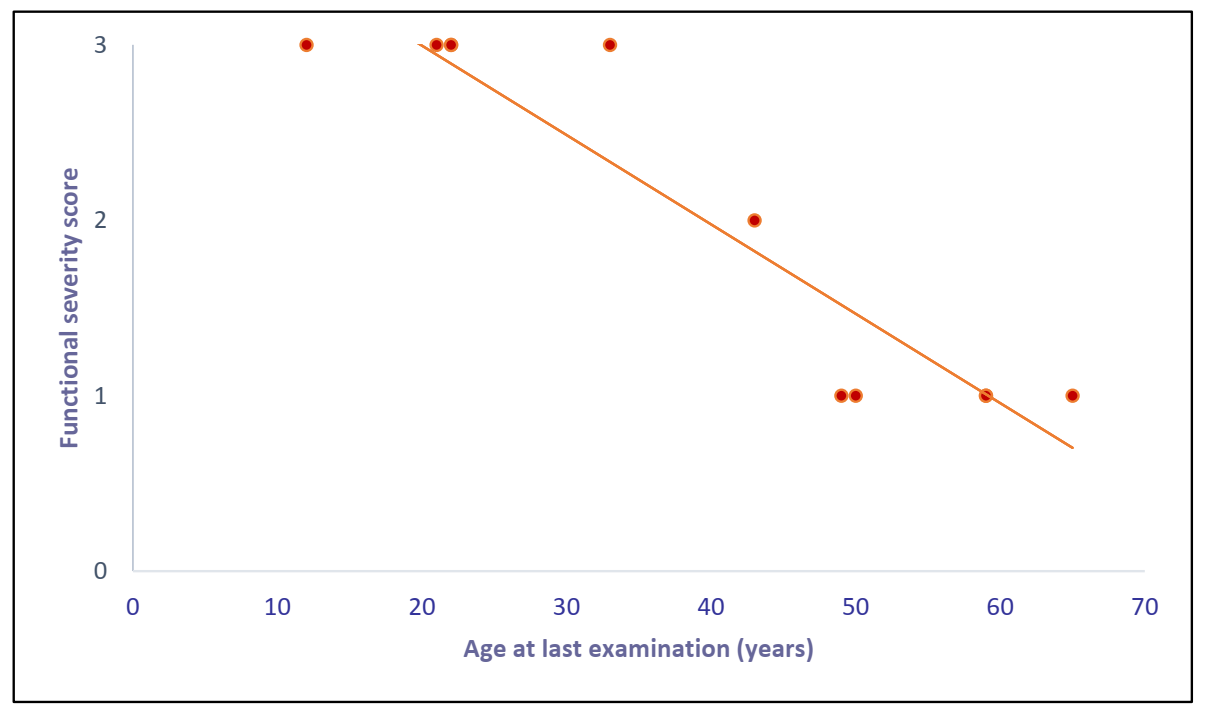
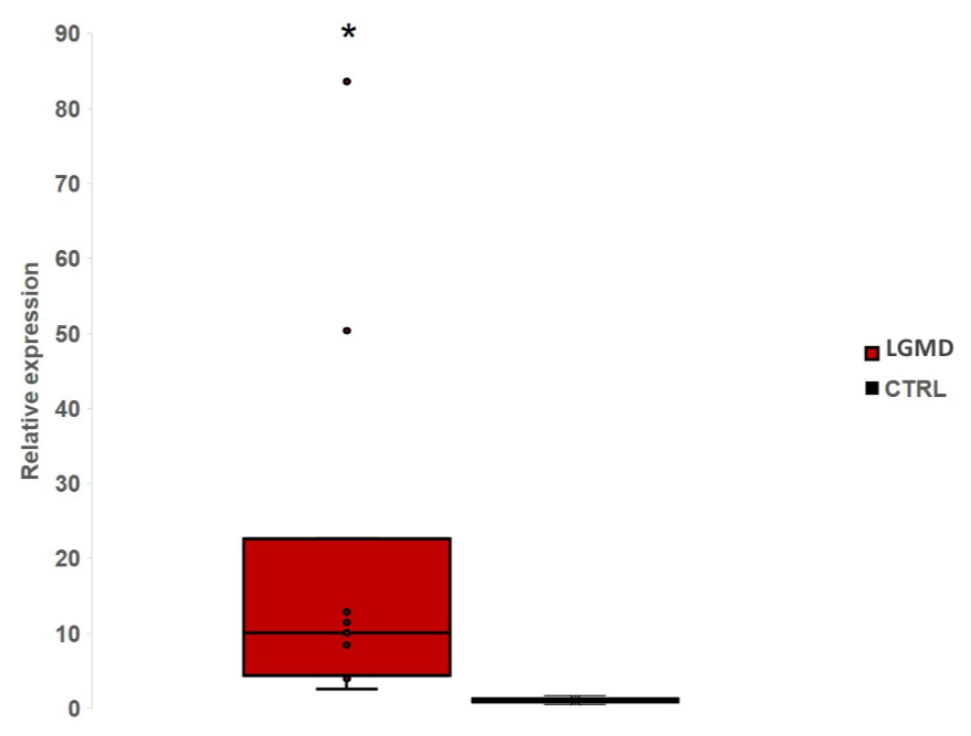
3.2. miR-206 in LGMD Patients
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Straub, V.; Murphy, A.; Udd, B. creat229 th ENMC international workshop: Limb girdle muscular dystrophies—Nomenclature and reformed classification, 17–19 March 2017, Naarden, The Netherlands. Neuromuscul. Disord. 2018, 17–19. [Google Scholar] [CrossRef]
- Murphy, A.P.; Straub, V. The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies. J. Neuromuscul. Dis. 2015, 2, S7–S19. [Google Scholar] [CrossRef]
- Pegoraro, E.; Hoffman, E.P. Limb-Girdle Muscular Dystrophy Overview. In Genereviews; University of Washington: Seattle, WA, USA, 2012. [Google Scholar]
- Bushby, K.M. Diagnosis and management of the limb girdle muscular dystrophies. Pract. Neurol 2009, 9, 314–323. [Google Scholar] [CrossRef]
- Torella, A.; Fanin, M.; Mutarelli, M.; Peterle, E.; Del Vecchio Blanco, F.; Rispoli, R.; Savarese, M.; Garofalo, A.; Piluso, G.; Morandi, L.; et al. Next-Generation Sequencing Identifies Transportin 3 as the Causative Gene for LGMD1F. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Cenacchi, G.; Papa, V.; Pegoraro, V.; Marozzo, R.; Fanin, M.; Angelini, C. Review: Danon disease: Review of natural history and recent advances. Neuropathol. Appl. Neurobiol. 2020, 46, 303–322. [Google Scholar] [CrossRef]
- Peterle, E.; Fanin, M.; Semplicini, C.; Padilla, J.J.V.; Nigro, V.; Angelini, C. Clinical phenotype, muscle MRI and muscle pathology of LGMD1F. J. Neurol. 2013, 260, 2033–2041. [Google Scholar] [CrossRef]
- Fanin, M.; Peterle, E.; Fritegotto, C.; Nascimbeni, A.C.; Tasca, E.; Torella, A.; Nigro, V.; Angelini, C. Incomplete penetrance in limb-girdle muscular dystrophy type 1F. Muscle Nerve 2015, 52, 305–306. [Google Scholar] [CrossRef]
- Angelini, C.; Pegoraro, V.C.G. The clinical and molecular spectrum of autosomal dominant limb-girdle muscular dystrophies focusing on transportinopathy. Expert Opin. Orphan Drugs 2019, 7, 223–232. [Google Scholar] [CrossRef]
- Politano, L.; Nigro, V.; Passamano, L.; Petretta, V.; Comi, L.I.; Papparella, S.; Nigro, G.; Rambaldi, P.F.; Raia, P.; Pini, A.; et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul. Disord. Nmd 2001, 11, 178–185. [Google Scholar] [CrossRef]
- Nigro, V.; Piluso, G. Spectrum of muscular dystrophies associated with sarcolemmal-protein genetic defects. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 585. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Defective assembly of sarcoglycan complex in patients with beta-sarcoglycan gene mutations. Study of aneural and innervated cultured myotubes. Neuropathol. Appl. Neurobiol. 2002, 28, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Fanin, M. Pathogenesis, clinical features and diagnosis of sarcoglycanopathies. Expert Opin. Orphan Drugs 2016, 4, 1239–1251. [Google Scholar] [CrossRef]
- Fayssoil, A.; Nardi, O.; Annane, D.; Orlikowski, D. Left ventricular function in alpha-sarcoglycanopathy and gamma-sarcoglycanopathy. Acta Neurol. Belg. 2014, 114, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Nardetto, L.; Borsato, C.; Padoan, R.; Fanin, M.; Nascimbeni, A.C.; Tasca, E. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B). Neurol. Res. 2010, 32, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Gläser, D.; Joshi, P.R.; Zierz, S.; Wenninger, S.; Schoser, B.; Deschauer, M. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J. Neurol. 2016, 263, 743–750. [Google Scholar] [CrossRef]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerve 2017, 55, 55–68. [Google Scholar] [CrossRef]
- Burch, P.M.; Pogoryelova, O.; Palandra, J.; Goldstein, R.; Bennett, D.; Fitz, L.; Guglieri, M.; Bettolo, C.M.; Straub, V.; Evangelista, T.; et al. Reduced serum myostatin concentrations associated with genetic muscle disease progression. J. Neurol. 2017, 264, 541–553. [Google Scholar] [CrossRef]
- Richard, I.; Brenguier, L.; Dinçer, P.; Roudaut, C.; Bady, B.; Burgunder, J.M.; Chemaly, R.; Garcia, C.A.; Halaby, G.; Jackson, C.E.; et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. Am. J. Hum. Genet. 1997, 60, 1128–1138. [Google Scholar]
- Lasa-Elgarresta, J.; Mosqueira-Martín, L.; Naldaiz-Gastesi, N.; Sáenz, A.; De Munain, A.L.; Vallejo-Illarramendi, A. Calcium Mechanisms in Limb-Girdle Muscular Dystrophy with CAPN3 Mutations. Int. J. Mol. Sci. 2019, 20, 4548. [Google Scholar] [CrossRef]
- Arikawa, E.; Hoffman, E.P.; Kaido, M.; Nonaka, I.; Sugita, H.; Arahata, K. The frequency of patients with dystrophin abnormalities in a limb-girdle patient population. Neurology 1991, 41, 1491. [Google Scholar] [CrossRef]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Fardeau, M.; Hillaire, D.; Mignard, C.; Feingold, N.; Feingold, J.; Mignard, D.; De Ubeda, B.; Collin, H.; Tomé, F.M.S.; Richard, I.; et al. Juvenile limb-girdle muscular dystrophy: Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 1996, 119, 295–308. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Benedicenti, F.; Fritegotto, C.; Nascimbeni, A.C.; Peterle, E.; Stanzial, F.; Cristofoletti, A.; Castellan, C.; Angelini, C. An intronic mutation causes severe LGMD2A in a large inbred family belonging to a genetic isolate in the Alps. Clin. Genet. 2012, 82, 601–602. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Nascimbeni, A.C.; Fulizio, L.; Angelini, C. The frequency of limb girdle muscular dystrophy 2A in northeastern Italy. Neuromuscul. Disord. Nmd 2005, 15, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Fanin, M.; Angelini, C. Protein and genetic diagnosis of limb girdle muscular dystrophy type 2A: The yield and the pitfalls. Muscle Nerve 2015, 52, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Horak, M.; Novak, J.; Bienertova-vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef]
- Zen, K.; Zhang, C.Y. Circulating microRNAs: A novel class of biomarkers to diagnose and monitor human cancers. Med. Res. Rev. 2012, 32, 326–348. [Google Scholar] [CrossRef]
- Pegoraro, V.; Missaglia, S.; Marozzo, R.; Tavian, D.; Angelini, C. MiRNAs as biomarkers of phenotype in neutral lipid storage disease with myopathy. Muscle Nerve 2020, 61, 253–257. [Google Scholar] [CrossRef]
- Missaglia, S.; Pegoraro, V.; Marozzo, R.; Tavian, D.; Angelini, C. Correlation between ETFDH mutations and dysregulation of serum myomiRs in MADD patients. Eur. J. Transl. Myol. 2020, 30, 20–25. [Google Scholar] [CrossRef]
- Angelini, C.; Fanin, M.; Freda, M.P.; Martinello, F.; Miorin, M.; Melacini, P.; Siciliano, G.; Pegoraro, E.; Rosa, M.; Danieli, G.A. Prognostic factors in mild dystrophinopathies. J. Neurol. Sci. 1996, 142, 70–78. [Google Scholar] [CrossRef]
- Mercuri, E.; Pichiecchio, A.; Counsell, S.; Allsop, J.; Cini, C.; Jungbluth, H.; Uggetti, C.; Bydder, G. A short protocol for muscle MRI in children with muscular dystrophies. Eur. J. Paediatr. Neurol. 2002, 6, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Williams, A.H.; Maxeiner, J.M.; Bezprozvannaya, S.; Shelton, J.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Investig. 2012, 122, 2054–2065. [Google Scholar] [CrossRef] [PubMed]
- Ma, G.; Wang, Y.; Li, Y.; Cui, L.; Zhao, Y.; Zhao, B.; Li, K. MiR-206, a key modulator of skeletal muscle development and disease. Int. J. Biol. Sci. 2015, 11, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Demonbreun, A.R.; McNally, E.M. Muscle cell communication in development and repair. Curr. Opin. Pharmacol. 2017, 34, 7–14. [Google Scholar] [CrossRef]
- Siracusa, J.; Koulmann, N.; Banzet, S. Circulating myomiRs: A new class of biomarkers to monitor skeletal muscle in physiology and medicine. J. Cachexia Sarcopenia Muscle 2018, 9, 20–27. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.-I.; Hashido, K. Three novel serum biomarkers, miR-1, miR-133a, and miR-206 for Limb-girdle muscular dystrophy, Facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ. Health Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef]
- Aguennouz, M.; Lo Giudice, C.; Licata, N.; Rodolico, C.; Musumeci, O.; Fanin, M.; Migliorato, A.; Ragusa, M.; Macaione, V.; Di Giorgio, R.M.; et al. MicroRNA signatures predict dysregulated vitamin D receptor and calcium pathways status in limb girdle muscle dystrophies (LGMD) 2A/2B. Cell Biochem. Funct. 2016, 34, 414–422. [Google Scholar] [CrossRef]
- Israeli, D.; Poupiot, J.; Amor, F.; Charton, K.; Lostal, W.; Jeanson-Leh, L.; Richard, I. Circulating miRNAs are generic and versatile therapeutic monitoring biomarkers in muscular dystrophies. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef]
- Marozzo, R.; Pegoraro, V.; Angelini, C. MiRNAs, Myostatin, and Muscle MRI Imaging as Biomarkers of Clinical Features in Becker Muscular Dystrophy. Diagnostics 2020, 10, 713. [Google Scholar] [CrossRef]
- Zaharieva, I.T.; Calissano, M.; Scoto, M.; Preston, M.; Cirak, S.; Feng, L.; Collins, J.; Kole, R.; Guglieri, M.; Straub, V.; et al. Dystromirs as serum biomarkers for monitoring the disease severity in Duchenne muscular Dystrophy. PLoS ONE 2013, 8, e80263. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. MiRNAs as serum biomarkers for Duchenne muscular dystrophy. Embo Mol. Med. 2011, 3, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Tao, Y.; Li, J.; Deng, Z.; Yan, Z.; Xiao, X.; Wang, D.Z. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. J. Cell Biol. 2010, 190, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, K.; Hagiwara, Y.; Ando, M.; Nakamura, A.; Takeda, S.; Hijikata, T. MicroRNA-206 is highly expressed in newly formed muscle fibers: Implications regarding potential for muscle regeneration and maturation in muscular dystrophy. Cell Struct. Funct. 2008, 33, 163–169. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Clinical and Molecular Features in Eleven LGMD Patients | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| pt. | Sex | Age at Last Examination | Onset | FSS | LGMD Subtype | Gene | Chromosome | cDNA Variant | Protein Amino-Acid Change |
| A | M | 12 | early | 3 | LGMDD2 | TNPO3 | 7q32.1-q32.2 | Frameshift mutation c.2767delC (exon 23) | p.Arg923AspfsTer17 |
| B | F | 43 | early | 2 | LGMDD2 | TNPO3 | 7q32.1-q32.2 | Frameshift mutation c.2767delC (exon 23) | p.Arg923AspfsTer17 |
| C | F | 33 | early | 3 | LGMDD2 | TNPO3 | 7q32.1 | Frameshift mutation c.2771delA (exon 22) | p.X924Cys |
| D | F | 59 | late | 1 | LGMDD2 | TNPO3 | 7q32.1 | Frameshift mutation c.2771delA (exon 22) | p.X924Cys |
| E | M | 22 | early adolescence | 3 | LGMDR1 | CAPN3 | 15q15.1-q21.1 | c.550delA (exon 4) Missense mutation c.1657G˃A (exon 13) | p.Thr184ArgfsTer36 p.Glu553Lys |
| F | F | 50 | late | 1 | LGMDR1 | CAPN3 | 15q15.1-q21.1 | Missense mutation c.1486G˃A (exon 11) | p.Gly496Arg |
| G | F | 59 | early | 1 | LGMDR1 | CAPN3 | 15q15.1 | Missense mutations c.202T˃C (exon 1) c.1061T˃C (exon 8) | p.Cys68Arg p.Val354Gly |
| H | M | 49 | late | 1 | LGMDR1 | CAPN3 | 15q15.1 | Missense mutation c.2242C˃G homozygous (exon 21) | p.Arg748Gly |
| I | F | 21 | early | 3 | LGMDR5 | SGCG | 13q12.12 | c.525delT homozygous | p.Phe175Leufs |
| L | F | 22 | early | 3 | LGMDR4 | SGCB | 4q12 | Nonsense mutation c.594T˃G c.418-425dup8bp | p.Gln142Lysfs24Ter |
| M | F | 65 | early adolescence | 1 | LGMDR3 | SGCA | 17q21.33 | Missense mutation c.850C˃T homozygous | p.Arg284Cys |
| MRC SCALE | ||||
|---|---|---|---|---|
| PATIENTS | QUADRICEPS | HAMSTRINGS | TIBIALIS ANTERIOR | GASTROCNEMIUS |
| A | 3/5sx 3/5dx | 3/5sx 3/5dx | 4/5sx 4/5dx | 4/5sx 4/5dx |
| B | 3/5sx 3/5dx | 4/5sx 4/5dx | 3/5sx 3/5dx | 2/5sx 2/5dx |
| C | 4/5sx 4/5dx | 3/5sx 4/5dx | 3/5sx 4/5dx | 2/5sx 2/5dx |
| D | 4/5sx 4/5dx | 4/5sx 4/5dx | 4/5sx 4/5dx | 4/5sx 4/5dx |
| E | 4/5sx 4/5dx | n.a. | 4/5sx 4/5dx | 4/5sx 4/5dx |
| F | 5/5sx 5/5dx | 3/5sx 3/5dx | 5/5sx 5/5dx | 5/5sx 5/5dx |
| G | 2/5sx 2/5dx | 2/5sx 2/5dx | 2/5sx 2/5dx | 2/5sx 2/5dx |
| H | 2/5sx 2/5dx | 2/5sx 2/5dx | 2/5sx 2/5dx | 2/5sx 2/5dx |
| I | 3+/5sx 3+/5dx | 3+/5sx 3+/5dx | 5/5 sx 4+/5 dx | 5/5sx 5/5dx |
| Mercuri Score in Muscle of Lgmd Patients | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thigh | Leg | |||||||||||||||||||||||||
| Patient | Rectus Femoris | Vastus Lateralis | Vastus Medialis | Sartorius | Biceps Femoris | Semimenbranosus/Semitendinosus | Adductor Magnus | Gracilis | Anterior Tibial | Extensor Longi Digitorum and Hallucis | Peroneus | Gastrocnemius | Soleus | |||||||||||||
| dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | dx | sx | |
| A | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 2a | 2a | 4 | 4 | 2a | 2a | 2a | 2a | 2a | 2a | 2a | 2a | 2b | 2b | 2a | 2a |
| B | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 2b | 2b | 4 | 4 | 3 | 3 | 2b | 2b | 2b | 2b | 2b | 2b | 4 | 4 | 3 | 3 |
| C | 2b | 2b | 2b | 2b | 2a | 2a | 3 | 3 | 2a | 2a | 2b | 2b | 2b | 2b | 3 | 3 | 2a | 2b | 2a | 2b | 2a | 2b | 4 | 4 | 2a | 2a |
| D | 3 | 4 | 2b | 2b | 2b | 2b | 4 | 4 | 2b | 2b | 2b | 2b | 2b | 2b | 3 | 3 | 2b | 2b | 2b | 2b | 2b | 2b | 4 | 4 | 2a | 2a |
| E | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 3 | 3 | 4 | 4 | 3 | 3 | 2a | 2a | 2a | 2a | 2b | 2b | 3 | 3 | 2a | 2a |
| F | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 2b | 2b | 3 | 3 | 3 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 0 | 0 |
| G | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 4 | 4 | 4 | 2b | 2b | 2a | 2b | 2a | 2a | 2b | 2b | 4 | 4 | 4 | 4 |
| H | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 4 | 4 | 4 | 4 | 4 | 4 | 3 | 3 | / | / | / | / | / | / | / | / | / | / |
| I | 2a | 2b | 2b | 2b | 3 | 3 | 2a | 2a | 3 | 3 | 4 | 4 | 1 | 1 | 1 | 1 | 2b | 2a | 2b | 2b | 2b | 2b | 1 | 1 | 2a | 2b |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pegoraro, V.; Angelini, C. Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies. Genes 2021, 12, 85. https://doi.org/10.3390/genes12010085
Pegoraro V, Angelini C. Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies. Genes. 2021; 12(1):85. https://doi.org/10.3390/genes12010085
Chicago/Turabian StylePegoraro, Valentina, and Corrado Angelini. 2021. "Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies" Genes 12, no. 1: 85. https://doi.org/10.3390/genes12010085
APA StylePegoraro, V., & Angelini, C. (2021). Circulating miR-206 as a Biomarker for Patients Affected by Severe Limb Girdle Muscle Dystrophies. Genes, 12(1), 85. https://doi.org/10.3390/genes12010085