Multi-Omics Approach Reveals miR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle

 ,
,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Phenotypic Data
2.2. DNA Extraction and Genotypic Data
2.3. Transcriptomic Data
2.3.1. mRNA Expression Data
2.3.2. miRNA Expression Data
2.4. Protein Data
2.5. Retrieval of SNPs in miRNA-Related Regions
2.6. Association Analysis
2.7. Pre-miRNA Secondary Stem–Loop Structures
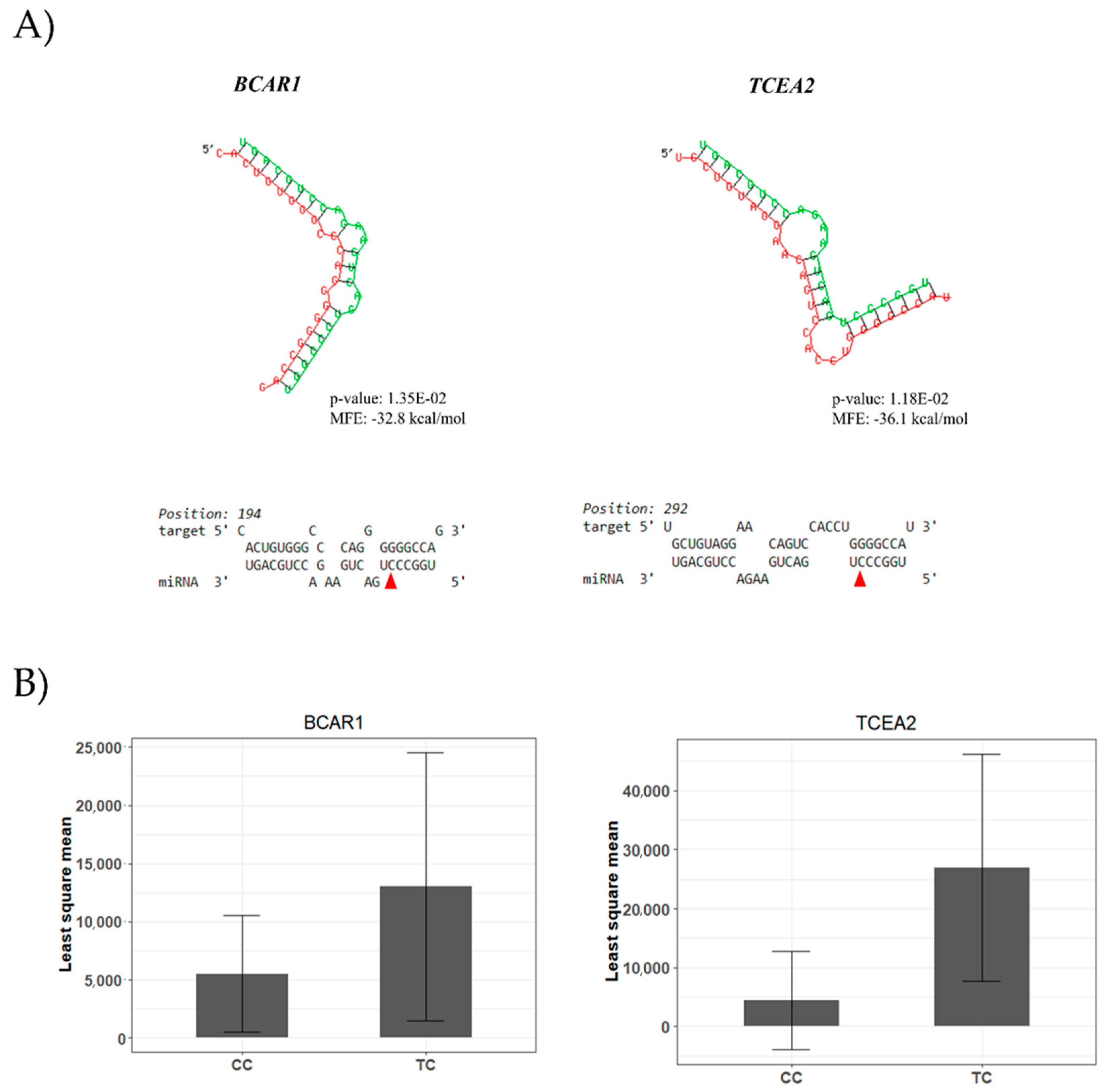
2.8. miRNA-mRNA Interaction Analysis
2.9. Pathway Analysis
3. Results
4. Discussion
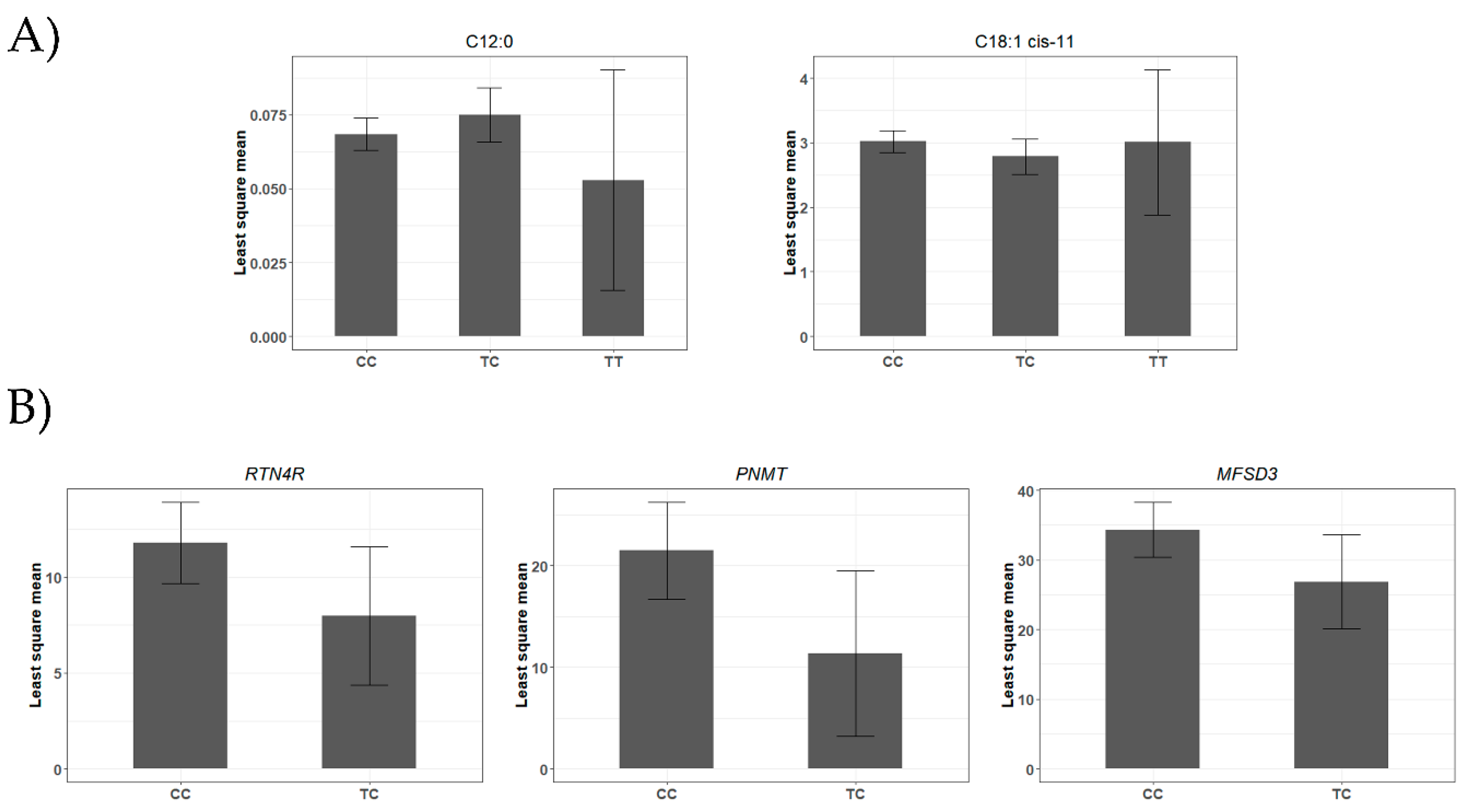
4.1. A miR-SNP Located in the bta-miR-2419-3p Sequence Was Associated with C12:0 and 18:1-cis 11 FAs Profiles
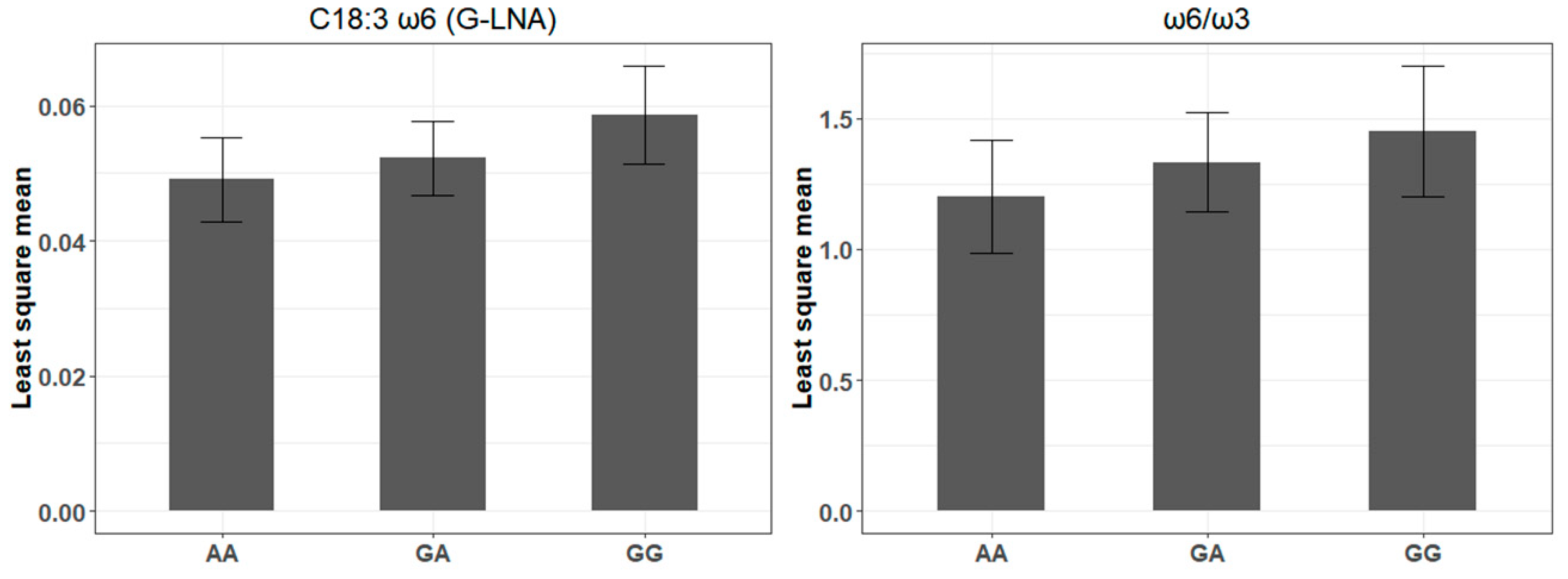
4.2. A bta-miR-193a-2-SNP May Influence γ-Linolenic Acid and ω6/ω3 Ratio Profiles
4.3. Seed SNP in the bta-miR-1291 Controlling the Composition of Many ω6-PUFA FAs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nakamura, M.T.; Nara, T.Y. Essential fatty acid synthesis and its regulation in mammals. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 145–150. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, X.; Huang, W.; Miao, X. Identification and characterization of differentially expressed miRNAs in subcutaneous adipose between Wagyu and Holstein cattle. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, P.S.N.; Coutinho, L.L.; Cesar, A.S.M.; Da Silva Diniz, W.J.; De Souza, M.M.; Andrade, B.G.; Koltes, J.E.; Mourão, G.B.; Zerlotini, A.; Reecy, J.M.; et al. Co-expression networks reveal potential regulatory roles of miRNAs in fatty acid composition of Nelore cattle. Front. Genet. 2019, 10, 651. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, L.L.; Matukumalli, L.K.; Sonstegard, T.S.; Van Tassell, C.P.; Gasbarre, L.C.; Capuco, A.V.; Smith, T.P.L. Discovery and profiling of bovine microRNAs from immune-related and embryonic tissues. Physiol. Genomics 2007, 29, 35–43. [Google Scholar] [CrossRef]
- Darnell, D.K.; Kaur, S.; Stanislaw, S.; Konieczka, J.K.; Yatskievych, T.A.; Antin, P.B. MicroRNA expression during chick embryo development. Dev. Dyn. 2006, 235, 3156–3165. [Google Scholar] [CrossRef]
- Hong, L.; Liu, R.; Qiao, X.; Wang, X.; Wang, S.; Li, J.; Wu, Z.; Zhang, H. Differential microRNAs expression in porcine endometrium involved in remodeling and angiogenesis that contribute to the embryonic implantation. Front. Genet. 2019, 10, 661. [Google Scholar] [CrossRef] [PubMed]
- Pokharel, K.; Peippo, J.; Honkatukia, M.; Seppälä, A.; Rautiainen, J.; Ghanem, N.; Hamama, T.M.; Crowe, M.A.; Andersson, M.; Li, M.H.; et al. Integrated ovarian mRNA and miRNA transcriptome profiling characterizes the genetic basis of prolificacy traits in sheep (Ovis aries). BMC Genomics 2018, 19, 1–17. [Google Scholar] [CrossRef]
- Cesar, A.S.M.; Regitano, L.C.A.; Poleti, M.D.; Andrade, S.C.S.; Tizioto, P.C.; Oliveira, P.S.N.; Felício, A.M.; do Nascimento, M.L.; Chaves, A.S.; Lanna, D.P.D.; et al. Differences in the skeletal muscle transcriptome profile associated with extreme values of fatty acids content. BMC Genomics 2016, 17, 961. [Google Scholar] [CrossRef]
- De Oliveira, P.S.N.; Coutinho, L.L.; Tizioto, P.C.; Cesar, A.S.M.; de Oliveira, G.B.; Diniz, W.J.D.S.; De Lima, A.O.; Reecy, J.M.; Mourão, G.B.; Zerlotini, A.; et al. An integrative transcriptome analysis indicates regulatory mRNA-miRNA networks for residual feed intake in Nelore cattle. Sci. Rep. 2018, 8, 17072. [Google Scholar] [CrossRef]
- Mukiibi, R.; Johnston, D.; Vinsky, M.; Fitzsimmons, C.; Stothard, P.; Waters, S.M.; Li, C. Bovine hepatic miRNAome profiling and differential miRNA expression analyses between beef steers with divergent feed efficiency phenotypes. Sci. Rep. 2020, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kappeler, B.I.G.; Regitano, L.C.A.; Poleti, M.D.; Cesar, A.S.M.; Moreira, G.C.M.; Gasparin, G.; Coutinho, L.L. MiRNAs differentially expressed in skeletal muscle of animals with divergent estimated breeding values for beef tenderness. BMC Mol. Biol. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Vignal, A.; Milan, D.; SanCristobal, M.; Eggen, A. A review on SNP and other types of molecular markers and their use in animal genetics. Genet. Sel. Evol. 2002, 34, 275–305. [Google Scholar] [CrossRef]
- Duan, R.; Pak, C.; Jin, P. Single nucleotide polymorphism associated with mature miR-125a alters the processing of pri-miRNA. Hum. Mol. Genet. 2007, 16, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Chen, L.; Luo, Z.; Zhang, T.; Chen, L.; Lou, P.; Sun, W.; Long, X.; Lan, J.; Wang, J.; et al. Spontaneous single nucleotide polymorphism in porcine microRNA-378 seed region leads to functional alteration. Biosci. Biotechnol. Biochem. 2018, 82, 1081–1089. [Google Scholar] [CrossRef]
- He, S.; Ou, H.; Zhao, C.; Zhang, J. Clustering Pattern and Functional Effect of SNPs in Human miRNA Seed Regions. Int J Genom. 2018. [Google Scholar] [CrossRef]
- Flynt, A.S.; Lai, E.C. Biological principles of microRNA-mediated regulation: Shared themes amid diversity. Nat. Rev. Genet. 2008, 9, 831–842. [Google Scholar] [CrossRef]
- Fang, L.; Sørensen, P.; Sahana, G.; Panitz, F.; Su, G.; Zhang, S.; Yu, Y.; Li, B.; Ma, L.; Liu, G.; et al. MicroRNA-guided prioritization of genome-wide association signals reveals the importance of microRNA-target gene networks for complex traits in cattle. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef]
- Jiang, Q.; Zhao, H.; Li, R.; Zhang, Y.; Liu, Y.; Wang, J.; Wang, X.; Ju, Z.; Liu, W.; Hou, M.; et al. In silico genome-wide miRNA-QTL-SNPs analyses identify a functional SNP associated with mastitis in Holsteins. BMC Genet. 2019, 20, 46. [Google Scholar] [CrossRef]
- Jevsinek Skok, D.; Godnic, I.; Zorc, M.; Horvat, S.; Dovc, P.; Kovac, M.; Kunej, T. Genome-wide in silico screening for microRNA genetic variability in livestock species. Anim. Genet. 2013, 44, 669–677. [Google Scholar] [CrossRef]
- Zorc, M.; Obsteter, J.; Dovc, P.; Kunej, T. Genetic Variability of MicroRNA Genes in 15 Animal Species. J. Genomics 2015, 3, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Tizioto, P.C.; Decker, J.E.; Taylor, J.F.; Schnabel, R.D.; Mudadu, M.A.; Silva, F.L.; Mourão, G.B.; Coutinho, L.L.; Tholon, P.; Sonstegard, T.S.; et al. Genome scan for meat quality traits in Nelore beef cattle. Physiol. Genomics 2013, 45, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Cesar, A.S.M.; Regitano, L.C.A.; Mourão, G.B.; Tullio, R.R.; Lanna, D.P.D.; Nassu, R.T.; Mudado, M.A.; Oliveira, P.S.N.; do Nascimento, M.L.; Chaves, A.S.; et al. Genome-wide association study for intramuscular fat deposition and composition in Nellore cattle. BMC Genet. 2014, 15, 39. [Google Scholar] [CrossRef] [PubMed]
- Hara, A.; Radin, N.S. Lipid extraction of tissues with a low-toxicity solvent. Anal. Biochem. 1978, 90, 420–426. [Google Scholar] [CrossRef]
- Christie, W.W. The Composition and Structure of Milk Lipids. In Developments in Dairy Chemistry, 2nd ed.; Springer: Amsterdam, The Netherlands, 1983; pp. 1–35. [Google Scholar]
- Tizioto, P.; Meirelles, S.; Tulio, R.; Rosa, A.; Alencar, M.; Medeiros, S.; Siqueira, F.; Feijó, G.; Silva, L.; Torres Junior, R.; et al. Candidate genes for production traits in Nelore beef cattle Candidate genes in Nelore. Genet. Mol. Res. 2012, 11, 4138–4144. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Loh, P.R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Das, S.; Forer, L.; Schönherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Diniz, W.J.S.; Mazzoni, G.; Coutinho, L.L.; Banerjee, P.; Geistlinger, L.; Cesar, A.S.M.; Bertolini, F.; Afonso, J.; De Oliveira, P.S.N.; Tizioto, P.C.; et al. Detection of co-expressed pathway modules associated with mineral concentration and meat quality in nelore cattle. Front. Genet. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Cesar, A.S.M.; Regitano, L.C.A.; Reecy, J.M.; Poleti, M.D.; Oliveira, P.S.N.; de Oliveira, G.B.; Moreira, G.C.M.; Mudadu, M.A.; Tizioto, P.C.; Koltes, J.E.; et al. Identification of putative regulatory regions and transcription factors associated with intramuscular fat content traits. BMC Genom. 2018, 19, 499. [Google Scholar] [CrossRef] [PubMed]
- Zhbannikov, I.Y.; Hunter, S.S.; Foster, J.A.; Settles, M.L. SeqyClean. In Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, Boston, MA, USA, 20–23 August 2017; pp. 407–416. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Tarazona, S.; Furió-Tarí, P.; Turrà, D.; Di Pietro, A.; Nueda, M.J.; Ferrer, A.; Conesa, A. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015, 43, e140. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.B.; Regitano, L.C.A.; Cesar, A.S.M.; Reecy, J.M.; Degaki, K.Y.; Poleti, M.D.; Felício, A.M.; Koltes, J.E.; Coutinho, L.L. Integrative analysis of microRNAs and mRNAs revealed regulation of composition and metabolism in Nelore cattle. BMC Genom. 2018, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Diniz, W.J.; Banerjee, P.; Mazzoni, G.; Coutinho, L.L.; Cesar, A.S.M.; Afonso, J.; Gromboni, C.F.; Nogueira, A.R.A.; Kadarmideen, H.N.; de Almeida Regitano, L.C. Interplay among miR-29 family, mineral metabolism, and gene regulation in Bos indicus muscle. Mol. Genet. Genomics 2020, 295, 1–15. [Google Scholar]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Poleti, M.D.; Regitano, L.C.A.; Souza, G.H.M.F.; Cesar, A.S.M.; Simas, R.C.; Silva-Vignato, B.; Montenegro, H.; Pértille, F.; Balieiro, J.C.C.; Cameron, L.C.; et al. Proteome alterations associated with the oleic acid and cis-9, trans-11 conjugated linoleic acid content in bovine skeletal muscle. J. Proteomics 2020, 222, 103792. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Olivova, P.; Daly, A.E.; Gebler, J.C. Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 2005, 28, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Gorenstein, M.V.; Li, G.Z.; Vissers, J.P.C.; Geromanos, S.J. Absolute quantification of proteins by LCMSE: A virtue of parallel MS acquisition. Mol. Cell. Proteomics 2006, 5, 144–156. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Sun, G.; Yan, J.; Noltner, K.; Feng, J.; Li, H.; Sarkis, D.A.; Sommer, S.S.; Rossi, J.J. SNPs in human miRNA genes affect biogenesis and function. RNA 2009, 15, 1640–1651. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Kruger, J.; Rehmsmeier, M. RNAhybrid: MicroRNA target prediction easy, fast and flexible. Nucleic Acids Res. 2006, 34, W451–W454. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Moszyńska, A.; Gebert, M.; Collawn, J.F.; Bartoszewski, R. SNPs in microRNA target sites and their potential role in human disease. Open Biol. 2017, 7, 170019. [Google Scholar] [CrossRef]
- Shi, J.; Sun, G. Effect of pre-miRNA-1658 gene polymorphism on chicken growth and carcass traits. Asian Australas. J. Anim. Sci. 2017, 30, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Burns, T.A.; Kadegowda, A.K.G.; Duckett, S.K.; Pratt, S.L.; Jenkins, T.C. Palmitoleic (16:1 cis-9) and cis-vaccenic (18:1 cis-11) acid alter lipogenesis in bovine adipocyte cultures. Lipids 2012, 47, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Kuhar, M.J.; Couceyro, P.R.; Lambert, P.D. Biosynthesis of Catecholamines. In Basic Neurochemistry; Lippnicott-Raven: Philadelphia, PA, USA, 1999. [Google Scholar]
- Sharara-Chami, R.I.; Zhou, Y.; Ebert, S.; Pacak, K.; Ozcan, U.; Majzoub, J.A. Epinephrine deficiency results in intact glucose counter-regulation, severe hepatic steatosis and possible defective autophagy in fasting mice. Int. J. Biochem. Cell Biol. 2012, 44, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.; Correia, G.; Coelho, M.; Araújo, J.R.; Pinho, M.J.; Teixeira, A.L.; Medeiros, R.; Ribeiro, L. Dietary unsaturated fatty acids differently affect catecholamine handling by adrenal chromaffin cells. J. Nutr. Biochem. 2015, 26, 563–570. [Google Scholar] [CrossRef] [PubMed]
- Lo Bianco, L.; Attrotto, M.T.; Torretta, S.; Masellis, R.; Rampino, A.; D’Ambrosio, E.; Di Giorgio, A.; Ferranti, L.; Fazio, L.; Gelao, B.; et al. Genetic variation is associated with RTN4R expression and working memory processing in healthy humans. Brain Res. Bull. 2017, 134, 162–167. [Google Scholar] [CrossRef]
- Schwab, M.E. Functions of Nogo proteins and their receptors in the nervous system. Nat. Rev. Neurosci. 2010, 11, 799–811. [Google Scholar] [CrossRef]
- Engelking, L.R. Fatty Acid Biosynthesis. In Textbook of Veterinary Physiological Chemistry; Elsevier: Amsterdam, The Netherlands, 2015; pp. 358–364. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Nomura, K.H.; Nomura, K. The acetyl-CoA transporter family SLC33. Mol. Aspects Med. 2013, 34, 586–589. [Google Scholar] [CrossRef]
- Dieterich, I.A.; Lawton, A.J.; Peng, Y.; Yu, Q.; Rhoads, T.W.; Overmyer, K.A.; Cui, Y.; Armstrong, E.A.; Howell, P.R.; Burhans, M.S.; et al. Acetyl-CoA flux regulates the proteome and acetyl-proteome to maintain intracellular metabolic crosstalk. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Palombo, V.; Milanesi, M.; Sgorlon, S.; Capomaccio, S.; Mele, M.; Nicolazzi, E.; Ajmone-Marsan, P.; Pilla, F.; Stefanon, B.; D’Andrea, M. Genome-wide association study of milk fatty acid composition in Italian Simmental and Italian Holstein cows using single nucleotide polymorphism arrays. J. Dairy Sci. 2018, 101, 11004–11019. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Kao, M.C.; Fang, J.Y.; Zouboulis, C.C.; Zhang, L.; Gallo, R.L.; Huang, C.M. Antimicrobial property of lauric acid against propionibacterium acnes: Its therapeutic potential for inflammatory acne vulgaris. J. Investig. Dermatol. 2009, 129, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Zentek, J.; Buchheit-Renko, S.; Ferrara, F.; Vahjen, W.; Van Kessel, A.G.; Pieper, R. Nutritional and physiological role of medium-chain triglycerides and medium-chain fatty acids in piglets. Anim. Health Res. Rev. 2011, 12, 83–93. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F.; DiNicolantonio, J.J. Lauric acid-rich medium-chain triglycerides can substitute for other oils in cooking applications and may have limited pathogenicity. Open Heart 2016, 3, e000467. [Google Scholar] [CrossRef] [PubMed]
- Titov, V.N.; Ivanov, G.A.; Antonov, A.M. Laurine fatty acids, medium fatty acids and triglycerides, hyperlipidemia, resistance to insulin, prevention of atherosclerosis and ateromatosis. Klin. Lab. Diagn. 2019, 64, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.H.; Joo, S.T. Fatty acid profiles of ten muscles from high and low marbled hanwoo steers. Korean J. Food Sci. Anim. Resour. 2016, 36, 679–688. [Google Scholar] [CrossRef]
- Arner, P.; Kulyté, A. MicroRNA regulatory networks in human adipose tissue and obesity. Nat. Rev. Endocrinol. 2015, 11, 276–288. [Google Scholar] [CrossRef]
- Zhang, Y.-Y.; Wang, H.-B.; Wang, Y.-N.; Wang, H.-C.; Zhang, S.; Hong, J.-Y.; Guo, H.-F.; Chen, D.; Yang, Y.; Zan, L.-S. Transcriptome analysis of mRNA and microRNAs in intramuscular fat tissues of castrated and intact male Chinese Qinchuan cattle. PLoS ONE 2017, 12, e0185961. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Chapkin, R.S. Importance of dietary γ-linolenic acid in human health and nutrition. J. Nutr. 1998, 128, 1411–1414. [Google Scholar] [CrossRef]
- Sergeant, S.; Rahbar, E.; Chilton, F.H. Gamma-linolenic acid, Dihommo-gamma linolenic, Eicosanoids and Inflammatory Processes. Eur. J. Pharmacol. 2016, 785, 77–86. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002, 56, 365–379. [Google Scholar] [CrossRef]
- Li, W.; Kuang, Z.; Zheng, M.; He, G.; Liu, Y. Multi-omics integrative analysis to access role of coiled-coil domain-containing 80 in lipid metabolism. Biochem. Biophys. Res. Commun. 2020, 526, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Mariman, E.C.M.; Wang, P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell. Mol. Life Sci. 2010, 67, 1277–1292. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ojeda, F.J.; Méndez-Gutiérrez, A.; Aguilera, C.M.; Plaza-Díaz, J. Extracellular matrix remodeling of adipose tissue in obesity and metabolic diseases. Int. J. Mol. Sci. 2019, 20, 4888. [Google Scholar] [CrossRef] [PubMed]
- Afonso, J.; Coutinho, L.L.; Tizioto, P.C.; da Silva Diniz, W.J.; de Lima, A.O.; Rocha, M.I.P.; Buss, C.E.; Andrade, B.G.N.; Piaya, O.; da Silva, J.V.; et al. Muscle transcriptome analysis reveals genes and metabolic pathways related to mineral concentration in Bos indicus. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Braud, M. Identification and Analysis of Polymorphisms in miRNA Genes and Associated Targets in Bos Taurus and Bos Primigenius for Genomic Selection in Cattle. Ph.D. Tesis, National University of Ireland, Galway, Ireland, 2017. [Google Scholar]
- Kim, B.; Nesvizhskii, A.I.; Rani, P.G.; Hahn, S.; Aebersold, R.; Ranish, J.A. The transcription elongation factor TFIIS is a component of RNA polymerase II preinitiation complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 16068–16073. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Roberts, M.A.; Hoff, B. How dietary arachidonic- and docosahexaenoic- acid rich oils differentially affect the murine hepatic transcriptome. Lipids Health Dis. 2006, 5, 10. [Google Scholar] [CrossRef]
- Saini, R.K.; Keum, Y.S. Omega-3 and omega-6 polyunsaturated fatty acids: Dietary sources, metabolism, and significance—A review. Life Sci. 2018, 203, 255–267. [Google Scholar] [CrossRef]
- Daetwyler, H.D.; Swan, A.A.; Van Der Werf, J.H.; Hayes, B.J. Accuracy of pedigree and genomic predictions of carcass and novel meat quality traits in multi-breed sheep data assessed by cross-validation. Genet. Sel. Evol. 2012, 44, 33. [Google Scholar] [CrossRef]
- Nagatake, T.; Kunisawa, J. Emerging roles of metabolites of ω3 and ω6 essential fatty acids in the control of intestinal inflammation. Int. Immunol. 2019, 31, 569–577. [Google Scholar] [CrossRef]
- Harvey, K.A.; Walker, C.L.; Pavlina, T.M.; Xu, Z.; Zaloga, G.P.; Siddiqui, R.A. Long-chain saturated fatty acids induce pro-inflammatory responses and impact endothelial cell growth. Clin. Nutr. 2010, 29, 492–500. [Google Scholar] [CrossRef]
- Hayes, K. Dietary fat and heart health: In search of the ideal fat. Asia Pac. J. Clin. Nutr. 2002, 11, S394–S400. [Google Scholar] [CrossRef]
- Simopoulos, A.P. The omega-6/omega-3 fatty acid ratio: Health implications. Oléagineux Corps Gras Lipides 2010, 17, 267–275. [Google Scholar] [CrossRef]
- Kumar, D.; Bansal, G.; Narang, A.; Basak, T.; Abbas, T.; Dash, D. Integrating transcriptome and proteome profiling: Strategies and applications. Proteomics 2016, 16, 2533–2544. [Google Scholar] [CrossRef] [PubMed]
- Greenbaum, D.; Colangelo, C.; Williams, K.; Gerstein, M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miR-SNP | Bp * | miRNA | MAF | Phenotype | β | SE | p-Value |
|---|---|---|---|---|---|---|---|
| rs43400521T>C | 13 | bta-miR-2419-3p | 0.07 | C12:0 | 8.16 × 10−3 | 3.41 × 10−3 | 1.68 × 10−2 |
| C18:1 cis-11 | −4.13 × 10−1 | 1.72 × 10−1 | 1.72 × 10−2 | ||||
| rs516857374A>G | 10 | bta-miR-193a-2 | 0.41 | C18:3 ω6 (G-LNA) | 7.28 × 10−3 | 2.03 × 10−3 | 5.27 × 10−4 |
| ω6/ω3 | 1.94 × 10−1 | 7.13 × 10−2 | 7.26 × 10−3 | ||||
| rs110817643C>T | 6 | bta-miR-1291 | 0.04 | C18:2 cis-9 cis-12 ω6 | −2.81 × 10−1 | 1.03 × 10−1 | 6.24 × 10−3 |
| C20:3 ω6 | −2.88 × 10−2 | 1.13 × 10−2 | 1.12 × 10−2 | ||||
| C20:4 ω6 | −9.44 × 10−2 | 3.18 × 10−2 | 3.10 × 10−3 | ||||
| PUFA | −5.05 × 10−1 | 1.67 × 10−1 | 2.61 × 10−3 | ||||
| PUFA:SFA | −1.25 × 10−2 | 3.97 × 10−3 | 1.79 × 10−3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, T.F.; Coutinho, L.L.; Bruscadin, J.J.; da Silva Diniz, W.J.; Petrini, J.; Andrade, B.G.N.; de Oliveira, P.S.N.; Poleti, M.D.; Cesar, A.S.M.; da Silveira, J.C.; et al. Multi-Omics Approach Reveals miR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle. Genes 2021, 12, 67. https://doi.org/10.3390/genes12010067
Cardoso TF, Coutinho LL, Bruscadin JJ, da Silva Diniz WJ, Petrini J, Andrade BGN, de Oliveira PSN, Poleti MD, Cesar ASM, da Silveira JC, et al. Multi-Omics Approach Reveals miR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle. Genes. 2021; 12(1):67. https://doi.org/10.3390/genes12010067
Chicago/Turabian StyleCardoso, Tainã Figueiredo, Luiz Lehmann Coutinho, Jennifer Jessica Bruscadin, Wellison Jarles da Silva Diniz, Juliana Petrini, Bruno Gabriel Nascimento Andrade, Priscila Silva Neubern de Oliveira, Mirele Daiana Poleti, Aline Silva Mello Cesar, Juliano Coelho da Silveira, and et al. 2021. "Multi-Omics Approach Reveals miR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle" Genes 12, no. 1: 67. https://doi.org/10.3390/genes12010067
APA StyleCardoso, T. F., Coutinho, L. L., Bruscadin, J. J., da Silva Diniz, W. J., Petrini, J., Andrade, B. G. N., de Oliveira, P. S. N., Poleti, M. D., Cesar, A. S. M., da Silveira, J. C., Chiaratti, M. R., Zerlotini, A., Mourão, G. B., & de Almeida Regitano, L. C. (2021). Multi-Omics Approach Reveals miR-SNPs Affecting Muscle Fatty Acids Profile in Nelore Cattle. Genes, 12(1), 67. https://doi.org/10.3390/genes12010067