Oxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
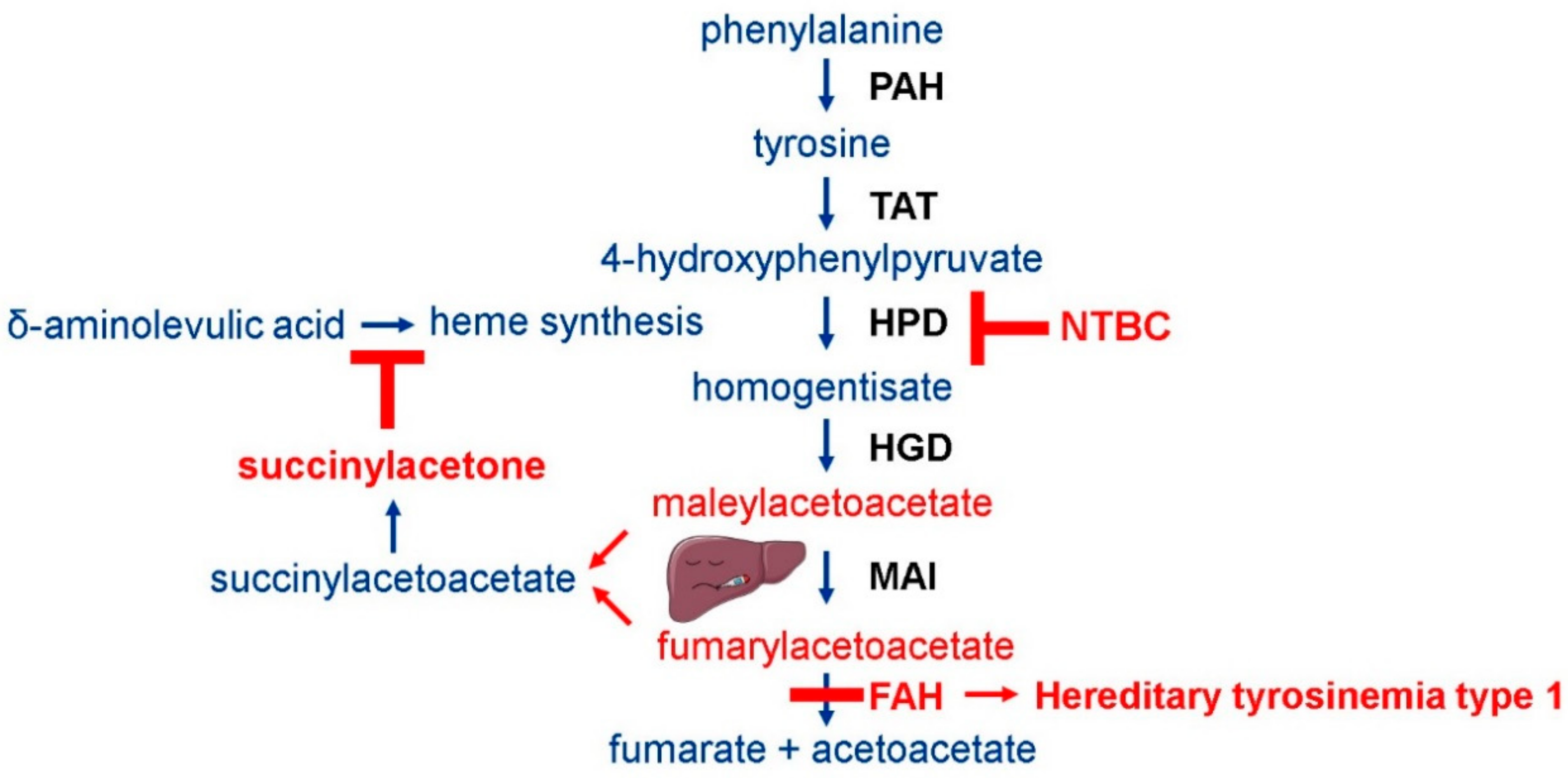
1. Introduction
2. Materials and Methods
2.1. Tyrosinemia Type 1 Mouse Model
2.2. Animal Experiments
2.3. Sample Collection and Preparation
2.4. Western Blot
2.5. Isolation of RNA and Reverse Transcriptase-Polymerase Chain Reaction (PCR)
2.6. Quantitative Real-Time PCR (qPCR)
2.7. Dried Blood SPOT (DBS) Analysis
2.8. Histopathology
2.9. Microarray Profiling
3. Results
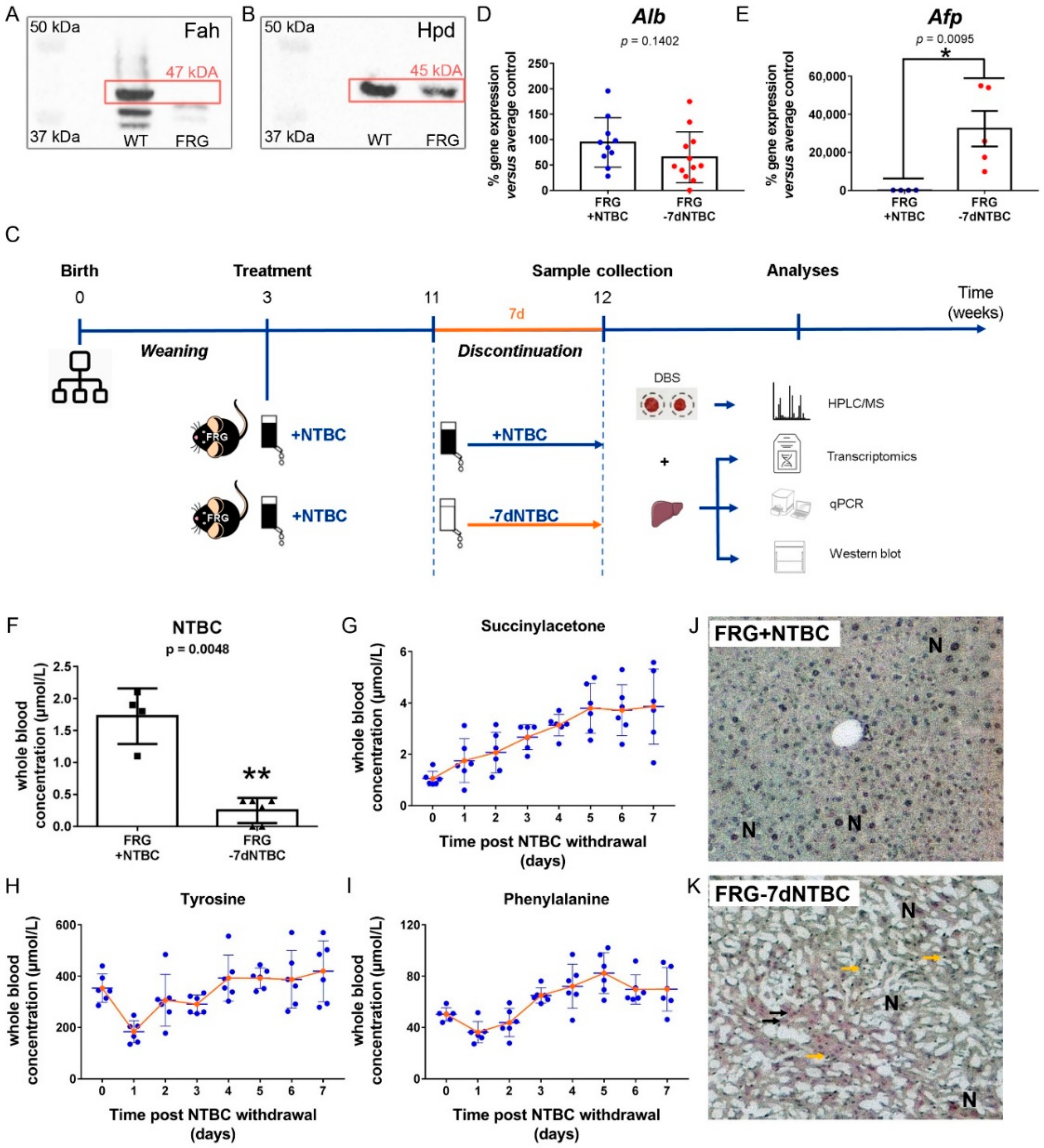
3.1. Short-Term NTBC Therapy Discontinuation Inflicts Severe Liver Damage in Fah-Deficient Mice
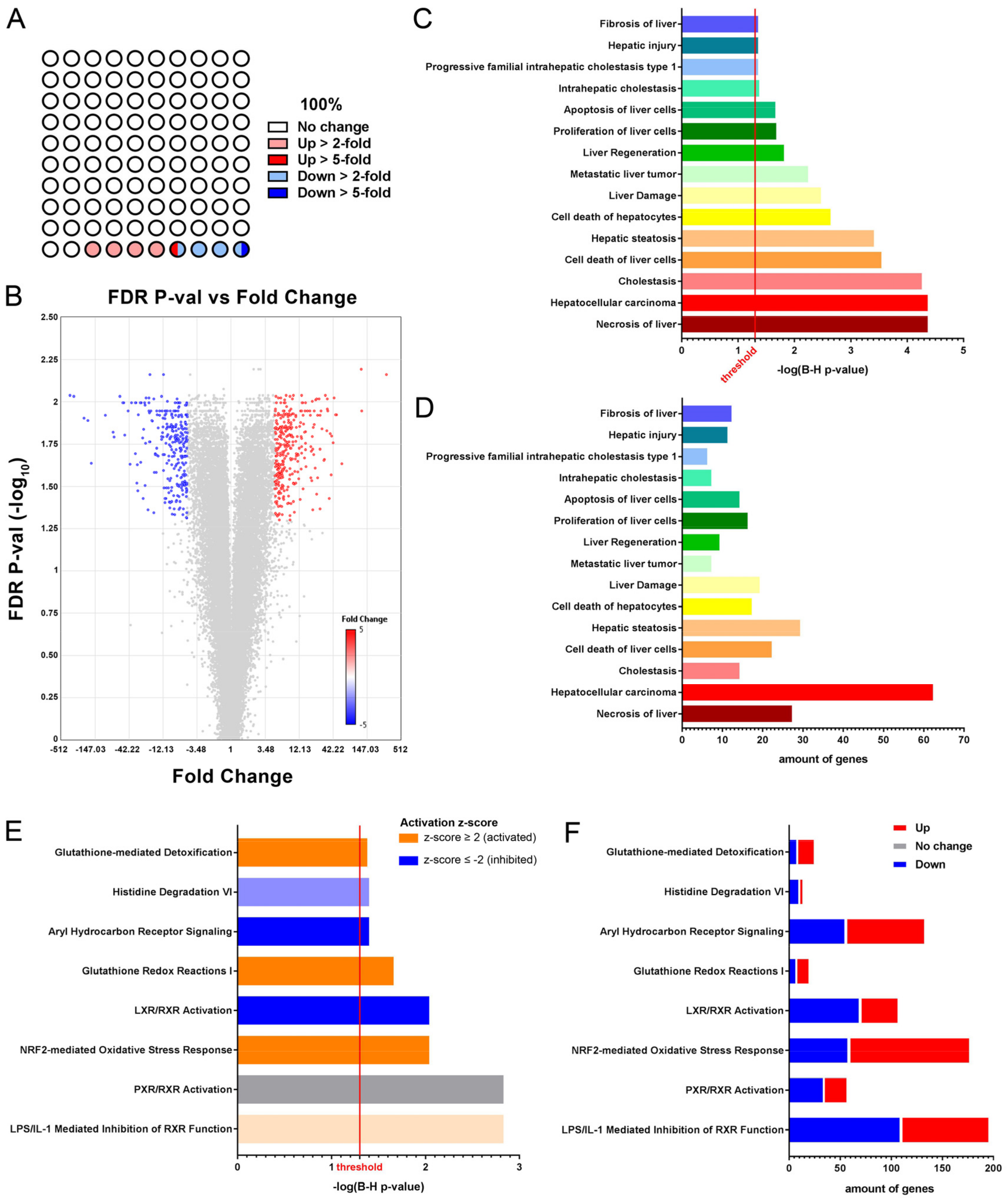
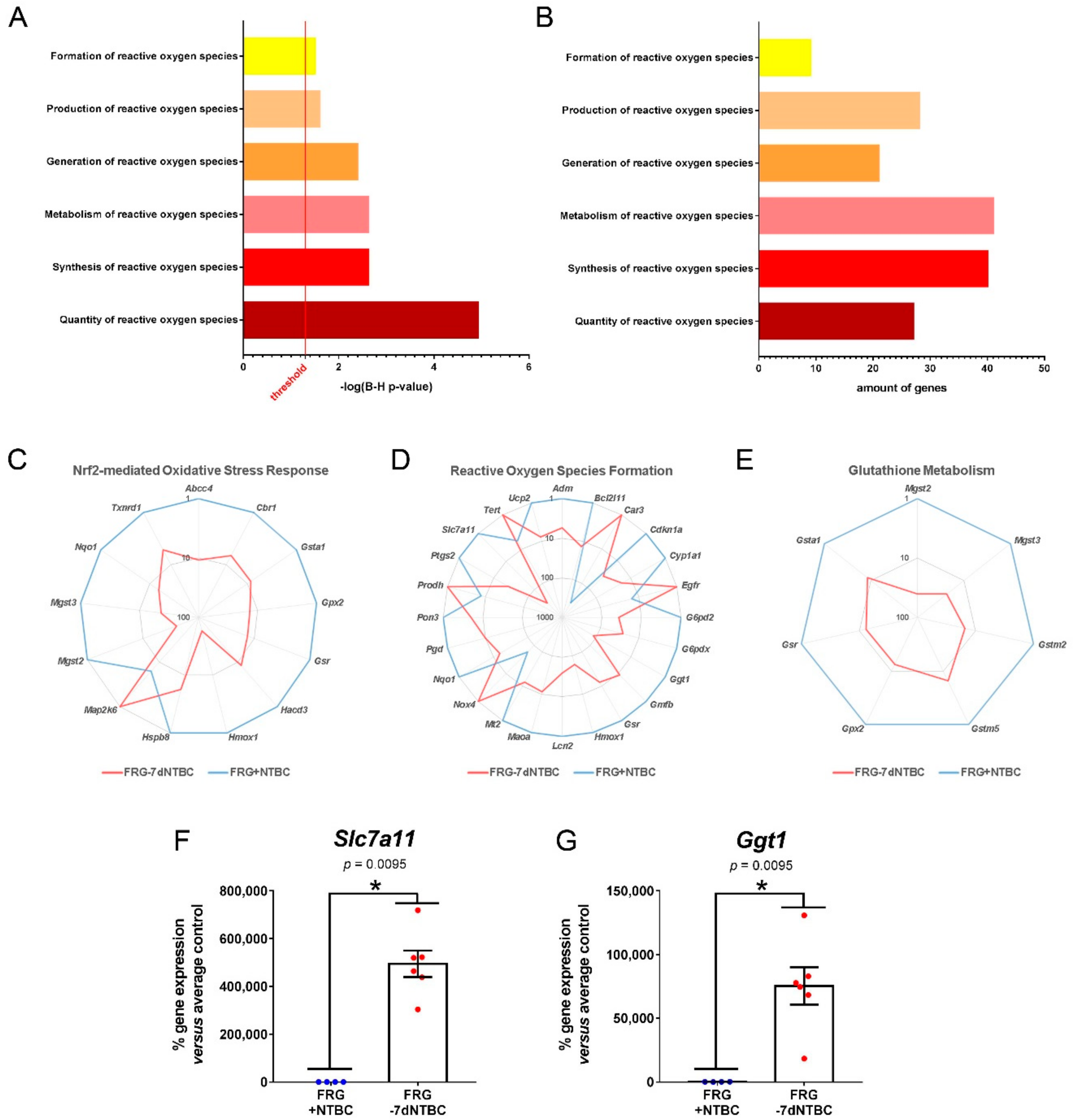
3.2. Glutathione Metabolism and Oxidative Stress Related Pathways Are Activated in Fah-Deficient Livers upon Short-Term NTBC Therapy Discontinuation
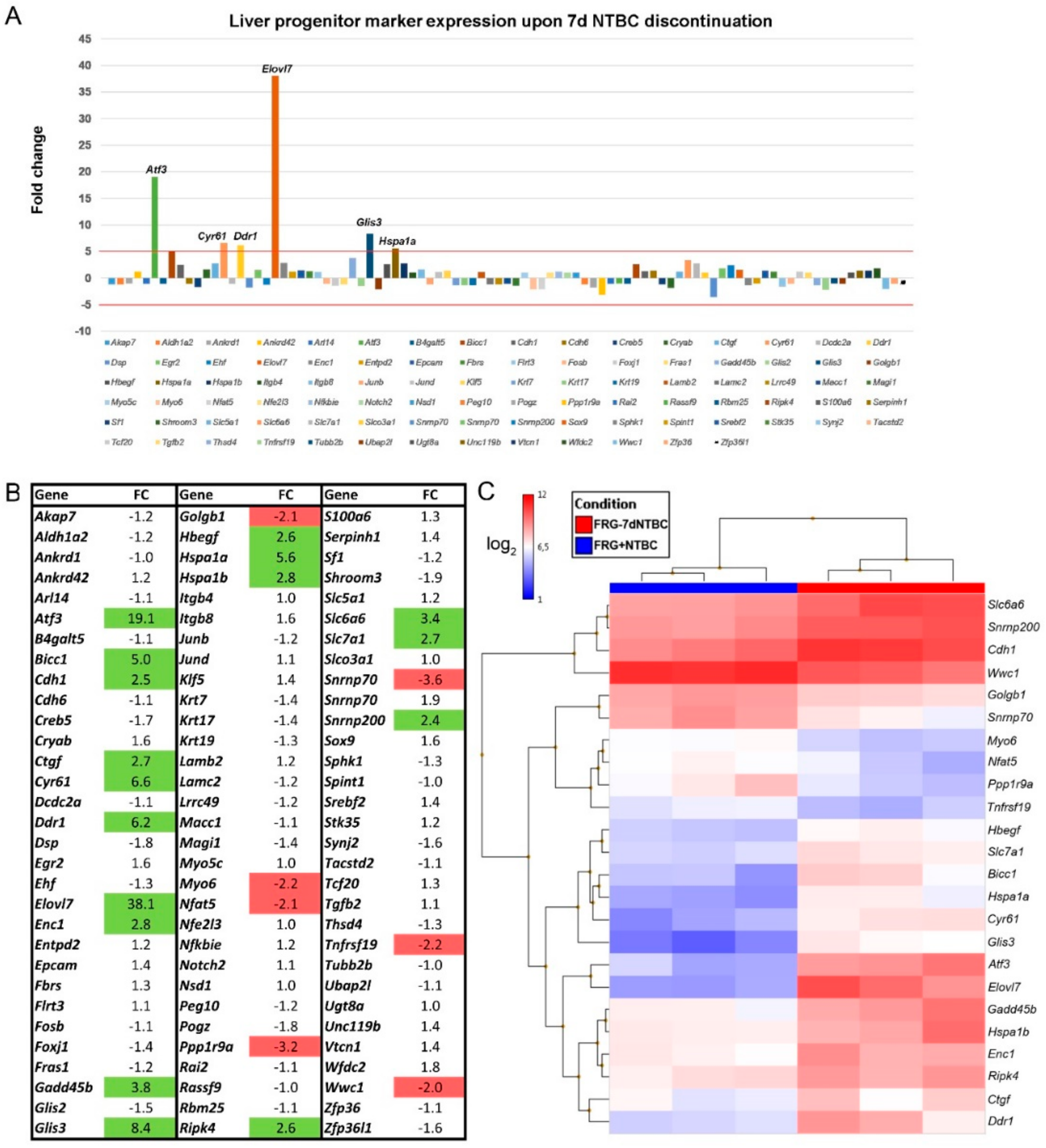
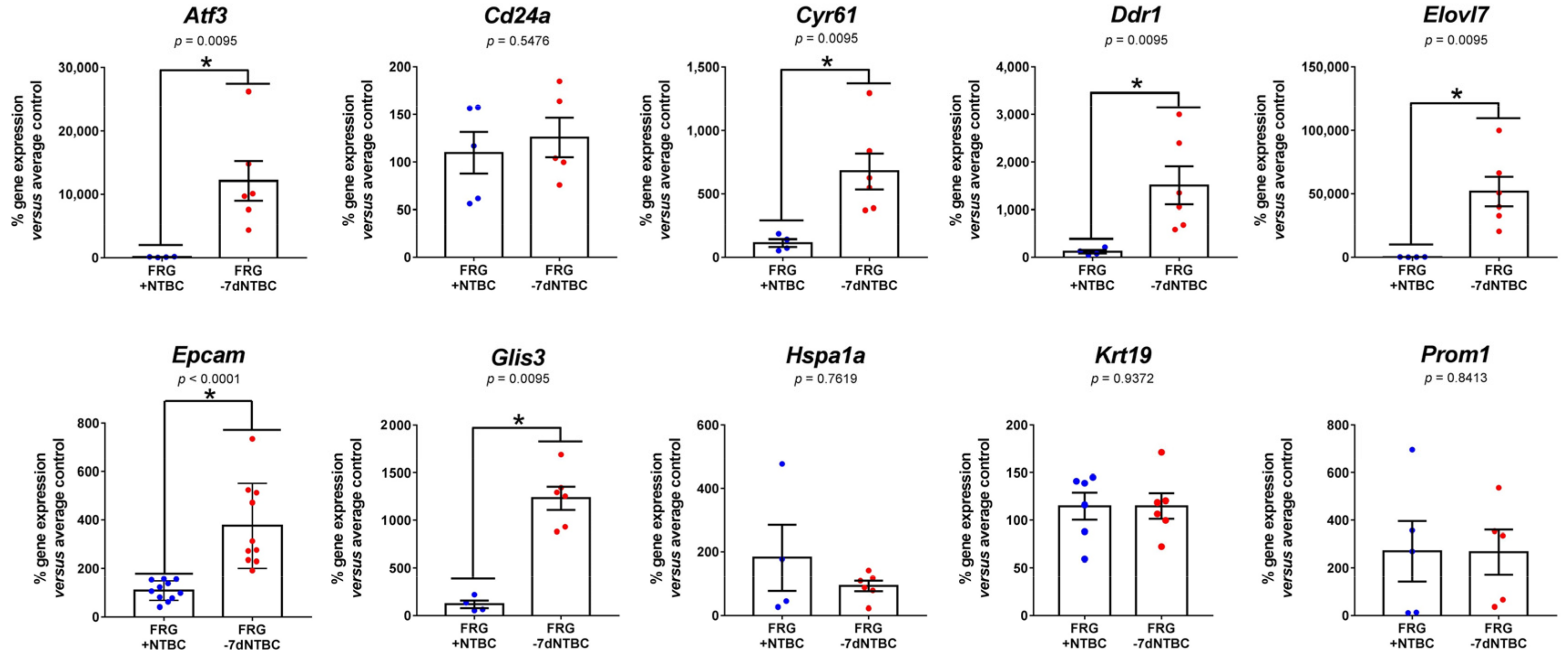
3.3. Fah-Deficient Liver Tissue Exhibits Increased Expression of Liver Progenitor Markers after Short-Term NTBC Therapy Discontinuation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- De Laet, C.; Dionisi-Vici, C.; Leonard, J.V.; McKiernan, P.; Mitchell, G.; Monti, L.; de Baulny, H.O.; Pintos-Morell, G.; Spiekerkötter, U. Recommendations for the management of tyrosinaemia type 1. Orphanet J. Rare Dis. 2013, 8, 8. [Google Scholar] [CrossRef]
- Alvarez, F.; Atkinson, S.; Bouchard, M.; Brunel-Guitton, C.; Buhas, D.; Bussières, J.F.; Dubois, J.; Fenyves, D.; Goodyer, P.; Gosselin, M.; et al. The Québec NTBC study. Adv. Exp. Med. Biol. 2017, 959, 187–195. [Google Scholar] [PubMed]
- Larochelle, J. Discovery of hereditary tyrosinemia in Saguenay-Lac St-Jean. Adv. Exp. Med. Biol. 2017, 959, 3–8. [Google Scholar]
- Äärelä, L.; Hiltunen, P.; Soini, T.; Vuorela, N.; Huhtala, H.; Nevalainen, P.I.; Heikinheimo, M.; Kivelä, L.; Kurppa, K. Type 1 tyrosinemia in Finland: A nationwide study. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bliksrud, Y.T.; Brodtkorb, E.; Backe, P.H.; Woldseth, B.; Rootwelt, H. Hereditary tyrosinaemia type 1 in Norway: Incidence and three novel small deletions in the fumarylacetoacetase gene. Scand. J. Clin. Lab. Invest. 2012, 72, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, G.A.; Yang, H. Remaining challenges in the treatment of tyrosinemia from the clinician’s viewpoint. Adv. Exp. Med. Biol. 2017, 959, 205–213. [Google Scholar]
- Phaneuf, D.; Labelle, Y.; Berube, D.; Arden, K.; Cavenee, W.; Gagne, R.; Tanguay, R.M. Cloning and expression of the cDNA encoding human fumarylacetoacetate hydrolase, the enzyme deficient in hereditary tyrosinemia: Assignment of the gene to chromosome 15. Am. J. Hum. Genet. 1991, 48, 525–535. [Google Scholar]
- Maiorana, A.; Dionisi-Vici, C. NTBC and correction of renal dysfunction. Adv. Exp. Med. Biol. 2017, 959, 93–100. [Google Scholar]
- Jorquera, R.; Tanguay, R.M. The mutagenicity of the tyrosine metabolite, fumarylacetoacetate, is enhanced by glutathione depletion. Biochem. Biophys. Res. Commun. 1997, 232, 42–48. [Google Scholar] [CrossRef]
- Dawson, C.; Ramachandran, R.; Safdar, S.; Murphy, E.; Swayne, O.; Katz, J.; Newsome, P.N.; Geberhiwot, T. Severe neurological crisis in adult patients with Tyrosinemia type 1. Ann. Clin. Transl. Neurol. 2020, 7, 1732–1737. [Google Scholar] [CrossRef]
- Morrow, G.; Tanguay, R.M. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv. Exp. Med. Biol. 2017, 959, 9–21. [Google Scholar] [PubMed]
- Michals, K.; Matolon, R.; Wong, P.W. Dietary treatment of tyrosinemia type I: Importance of methionine restriction. J. Am. Diet. Assoc. 1978, 73, 507–514. [Google Scholar] [PubMed]
- Van Spronsen, F.J.; van Rijn, M.; Meyer, U.; Das, A.M. Dietary considerations in tyrosinemia type I. Adv. Exp. Med. Biol. 2017, 959, 197–204. [Google Scholar] [PubMed]
- McKiernan, P. Liver transplantation for hereditary tyrosinaemia type 1 in the United Kingdom. Adv. Exp. Med. Biol. 2017, 959, 85–91. [Google Scholar] [PubMed]
- Liu, Y.; Luo, Y.; Xia, L.; Qiu, B.J.; Zhou, T.; Feng, M.X.; Xue, F.; Chen, X.S.; Han, L.S.; Zhang, J.J.; et al. Living-donor liver transplantation for children with tyrosinemia type I. J. Dig. Dis. 2020, 21, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Karaca, C.A.; Yilmaz, C.; Farajov, R.; Iakobadze, Z.; Aydogdu, S.; Kilic, M. Live donor liver transplantation for type 1 tyrosinemia: An analysis of 15 patients. Pediatr. Transplant. 2019, 23, e13498. [Google Scholar] [CrossRef]
- Lindstedt, S.; Holme, E.; Lock, E.A.; Hjalmarson, O.; Strandvik, B. Treatment of hereditary tyrosinaemia type I by inhibition of 4-hydroxyphenylpyruvate dioxygenase. Lancet 1992, 340, 813–817. [Google Scholar] [CrossRef]
- Neuckermans, J.; Mertens, A.; De Win, D.; Schwaneberg, U.; De Kock, J. A robust bacterial assay for high-throughput screening of human 4-hydroxyphenylpyruvate dioxygenase inhibitors. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Larochelle, J.; Alvarez, F.; Bussières, J.F.; Chevalier, I.; Dallaire, L.; Dubois, J.; Faucher, F.; Fenyves, D.; Goodyer, P.; Grenier, A.; et al. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol. Genet. Metab. 2012, 107, 49–54. [Google Scholar] [CrossRef]
- Gertsman, I.; Barshop, B.A.; Panyard-Davis, J.; Gangoiti, J.A.; Nyhan, W.L. Metabolic effects of increasing doses of nitisinone in the treatment of alkaptonuria. JIMD Rep. 2015, 24, 13–20. [Google Scholar]
- Thimm, E.; Herebian, D.; Assmann, B.; Klee, D.; Mayatepek, E.; Spiekerkoetter, U. Increase of CSF tyrosine and impaired serotonin turnover in tyrosinemia type I. Mol. Genet. Metab. 2011, 102, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.S.; Harrold, J.A.; Hughes, G.; Norman, B.P.; Devine, J.; Usher, J.; Hughes, A.T.; Khedr, M.; Gallagher, J.A.; Milan, A.M.; et al. Clinical and biochemical assessment of depressive symptoms in patients with Alkaptonuria before and after two years of treatment with nitisinone. Mol. Genet. Metab. 2018, 125, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Gertsman, I.; Gangoiti, J.A.; Nyhan, W.L.; Barshop, B.A. Perturbations of tyrosine metabolism promote the indolepyruvate pathway via tryptophan in host and microbiome. Mol. Genet. Metab. 2015, 114, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Van Ginkel, W.G.; Jahja, R.; Huijbregts, S.C.J.; Daly, A.; MacDonald, A.; De Laet, C.; Cassiman, D.; Eyskens, F.; Körver-Keularts, I.M.L.W.; Goyens, P.J.; et al. Neurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls. Orphanet J. Rare Dis. 2016, 11, 1–9. [Google Scholar] [CrossRef]
- García, M.I.; de la Parra, A.; Arias, C.; Arredondo, M.; Cabello, J.F. Long-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet. Mol. Genet. Metab. Rep. 2017, 11, 12–16. [Google Scholar] [CrossRef]
- Tsai, C.P.; Lin, P.Y.; Lee, N.C.; Niu, D.M.; Lee, S.M.; Hsu, W.M. Corneal lesion as the initial manifestation of tyrosinemia type II. J. Chin. Med. Assoc. 2006, 69, 286–288. [Google Scholar] [CrossRef]
- Stewart, R.M.K.; Briggs, M.C.; Jarvis, J.C.; Gallagher, J.A.; Ranganath, L. Reversible keratopathy due to hypertyrosinaemia following intermittent low-dose nitisinone in alkaptonuria: A case report. JIMD Rep. 2014, 17, 1–6. [Google Scholar]
- Azuma, H.; Paulk, N.; Ranade, A.; Dorrell, C.; Al-Dhalimy, M.; Ellis, E.; Strom, S.; Kay, M.A.; Finegold, M.; Grompe, M. Robust expansion of human hepatocytes in Fah-/-/Rag2-/-/Il2rg-/- mice. Nat. Biotechnol. 2007, 25, 903–910. [Google Scholar] [CrossRef]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. BMJ Open Sci. 2020, 4, e100115. [Google Scholar] [CrossRef]
- Verhulst, S.; Roskams, T.; Sancho-Bru, P.; van Grunsven, L.A. Meta-Analysis of Human and Mouse Biliary Epithelial Cell Gene Profiles. Cells 2019, 8, 1117. [Google Scholar] [CrossRef]
- Masurel-Paulet, A.; Poggi-Bach, J.; Rolland, M.O.; Bernard, O.; Guffon, N.; Dobbelaere, D.; Sarles, J.; Ogier de Baulny, H.; Touati, G. NTBC treatment in tyrosinaemia type I: Long-term outcome in French patients. J. Inherit. Metab. Dis. 2008, 31, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Aktuglu Zeybek, A.C.; Kiykim, E.; Soyucen, E.; Cansever, S.; Altay, S.; Zubarioglu, T.; Erkan, T.; Aydin, A. Hereditary tyrosinemia type 1 in Turkey: Twenty year single-center experience. Pediatr. Int. 2015, 57, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.L.; Dalmau, J.; Del Toro, M.; Pintos-Morell, G.; Aldámiz-Echevarría, L. Tyrosinemia type 1 in Spain: Mutational analysis, treatment and long-term outcome. Pediatr. Int. 2011, 53, 985–989. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.E.S.; Dutra, F.; Soares, C.O.; Alves, A.N.L.; Bevilacqua, E.; Gagioti, S.M.; Penatti, C.A.A.; Bechara, E.J.H. Liver damage induced by succinylacetone: A shared redox imbalance mechanism between tyrosinemia and hepatic porphyrias. J. Braz. Chem. Soc. 2017, 28, 1297–1307. [Google Scholar] [CrossRef]
- Lock, E.A.; Gaskin, P.; Ellis, M.K.; McLean Provan, W.; Robinson, M.; Smith, L.L. Tissue distribution of 2-(2-nitro-4-trifluoromethylbenzoyl)-cyclohexane-1,3-dione (NTBC) and its effect on enzymes involved in tyrosine catabolism in the mouse. Toxicology 2000, 144, 179–187. [Google Scholar] [CrossRef]
- Alobaidy, H.; Barkaoui, E. Experience of a single center in NTBC use in management of hereditary tyrosinemia type I in Libya. Iran. J. Pediatr. 2015, 25, e3608. [Google Scholar] [CrossRef]
- Lewerenz, J.; Hewett, S.J.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The cystine/glutamate antiporter system xc- in health and disease: From molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 522–555. [Google Scholar] [CrossRef]
- Wang, A.W.; Wangensteen, K.J.; Wang, Y.J.; Zahm, A.M.; Moss, N.G.; Erez, N.; Kaestner, K.H. TRAP-seq identifies cystine/glutamate antiporter as a driver of recovery from liver injury. J. Clin. Investig. 2018, 128, 2297–2309. [Google Scholar] [CrossRef]
- Hsu, J.C.; Laz, T.; Mohn, K.L.; Taub, R. Identification of LRF-1, a leucine-zipper protein that is rapidly and highly induced in regenerating liver. Proc. Natl. Acad. Sci. USA 1991, 88, 3511–3515. [Google Scholar] [CrossRef]
- Weir, E.; Chen, Q.; Defrances, M.C.; Bell, A.; Taub, R.; Zarnegar, R. Rapid induction of mRNAs for liver regeneration factor and insulin-like growth factor binding protein-1 in primary cultures of rat hepatocytes by hepatocyte growth factor and epidermal growth factor. Hepatology 1994, 20, 955–960. [Google Scholar] [CrossRef]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Nobori, K.; Ito, H.; Tamamori-Adachi, M.; Adachi, S.; Ono, Y.; Kawauchi, J.; Kitajima, S.; Marumo, F.; Isobe, M. ATF3 inhibits doxorubicin-induced apoptosis in cardiac myocytes: A novel cardioprotective role of ATF3. J. Mol. Cell. Cardiol. 2002, 34, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Allan, A.L.; Albanese, C.; Pestell, R.G.; LaMarre, J. Activating Transcription Factor 3 Induces DNA Synthesis and Expression of Cyclin D1 in Hepatocytes. J. Biol. Chem. 2001, 276, 27272–27280. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; DeWille, J.W.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Jun, J.I.; Lau, L.F. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 945–963. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B. Discoidin domain receptor functions in physiological and pathological conditions. Int. Rev. Cell Mol. Biol. 2014, 310, 39–87. [Google Scholar]
- Moll, S.; Desmoulière, A.; Moeller, M.J.; Pache, J.C.; Badi, L.; Arcadu, F.; Richter, H.; Satz, A.; Uhles, S.; Cavalli, A.; et al. DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118474. [Google Scholar] [CrossRef]
- Kiamehr, M.; Heiskanen, L.; Laufer, T.; Düsterloh, A.; Kahraman, M.; Käkelä, R.; Laaksonen, R.; Aalto-Setälä, K. Dedifferentiation of primary hepatocytes is accompanied with reorganization of lipid metabolism indicated by altered molecular lipid and miRNA profiles. Int. J. Mol. Sci. 2019, 20, 2910. [Google Scholar] [CrossRef]
- Yoon, S.M.; Gerasimidou, D.; Kuwahara, R.; Hytiroglou, P.; Yoo, J.E.; Park, Y.N.; Theise, N.D. Epithelial cell adhesion molecule (EpCAM) marks hepatocytes newly derived from stem/progenitor cells in humans. Hepatology 2011, 53, 964–973. [Google Scholar] [CrossRef]
- Safarikia, S.; Carpino, G.; Overi, D.; Cardinale, V.; Venere, R.; Franchitto, A.; Onori, P.; Alvaro, D.; Gaudio, E. Distinct EpCAM-Positive Stem Cell Niches Are Engaged in Chronic and Neoplastic Liver Diseases. Front. Med. 2020, 7, 479. [Google Scholar] [CrossRef]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-Positive Hepatocellular Carcinoma Cells Are Tumor-Initiating Cells with Stem/Progenitor Cell Features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, P.; Habeb, A.M.; Garbuz, F.; Millward, A.; Wallis, S.; Moussa, K.; Akcay, T.; Taha, D.; Hogue, J.; Slavotinek, A.; et al. Expanding the clinical spectrum associated with GLIS3 mutations. J. Clin. Endocrinol. Metab. 2015, 100, E1362–E1369. [Google Scholar] [CrossRef] [PubMed]
- Wolf, J.H.; Bhatti, T.R.; Fouraschen, S.; Chakravorty, S.; Wang, L.; Kurian, S.; Salomon, D.; Olthoff, K.M.; Hancock, W.W.; Levine, M.H. Heat shock protein 70 is required for optimal liver regeneration after partial hepatectomy in mice. Liver Transpl. 2014, 20, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, A.; Limonciel, A.; Aschauer, L.; Moenks, K.; Bielow, C.; Leonard, M.O.; Hamon, J.; Carpi, D.; Ruzek, S.; Handler, A.; et al. Application of integrated transcriptomic, proteomic and metabolomic profiling for the delineation of mechanisms of drug induced cell stress. J. Proteom. 2013, 79, 180–194. [Google Scholar] [CrossRef]
- Lin, S.; Qiao, N.; Chen, H.; Tang, Z.; Han, Q.; Mehmood, K.; Fazlani, S.A.; Hameed, S.; Li, Y.; Zhang, H. Integration of transcriptomic and metabolomic data reveals metabolic pathway alteration in mouse spermatogonia with the effect of copper exposure. Chemosphere 2020, 256, 126974. [Google Scholar] [CrossRef]
- Zhu, M.; Li, L.; Lu, T.; Yoo, H.; Zhu, J.; Gopal, P.; Wang, S.C.; Porembka, M.R.; Rich, N.E.; Kagan, S.; et al. Uncovering Biological Factors That Regulate Hepatocellular Carcinoma Growth Using Patient-Derived Xenograft Assays. Hepatology 2020, 72, 1085–1101. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colemonts-Vroninks, H.; Neuckermans, J.; Marcelis, L.; Claes, P.; Branson, S.; Casimir, G.; Goyens, P.; Martens, G.A.; Vanhaecke, T.; De Kock, J. Oxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation. Genes 2021, 12, 3. https://doi.org/10.3390/genes12010003
Colemonts-Vroninks H, Neuckermans J, Marcelis L, Claes P, Branson S, Casimir G, Goyens P, Martens GA, Vanhaecke T, De Kock J. Oxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation. Genes. 2021; 12(1):3. https://doi.org/10.3390/genes12010003
Chicago/Turabian StyleColemonts-Vroninks, Haaike, Jessie Neuckermans, Lionel Marcelis, Paul Claes, Steven Branson, Georges Casimir, Philippe Goyens, Geert A. Martens, Tamara Vanhaecke, and Joery De Kock. 2021. "Oxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation" Genes 12, no. 1: 3. https://doi.org/10.3390/genes12010003
APA StyleColemonts-Vroninks, H., Neuckermans, J., Marcelis, L., Claes, P., Branson, S., Casimir, G., Goyens, P., Martens, G. A., Vanhaecke, T., & De Kock, J. (2021). Oxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation. Genes, 12(1), 3. https://doi.org/10.3390/genes12010003