Newly Emerged Serotype 1c of Shigella flexneri: Multiple Origins and Changing Drug Resistance Landscape


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains
2.2. DNA Preparation and Sequencing
2.3. Bioinformatics Analysis
2.4. Antimicrobial-Resistant Genes and Susceptibility Testing
3. Results
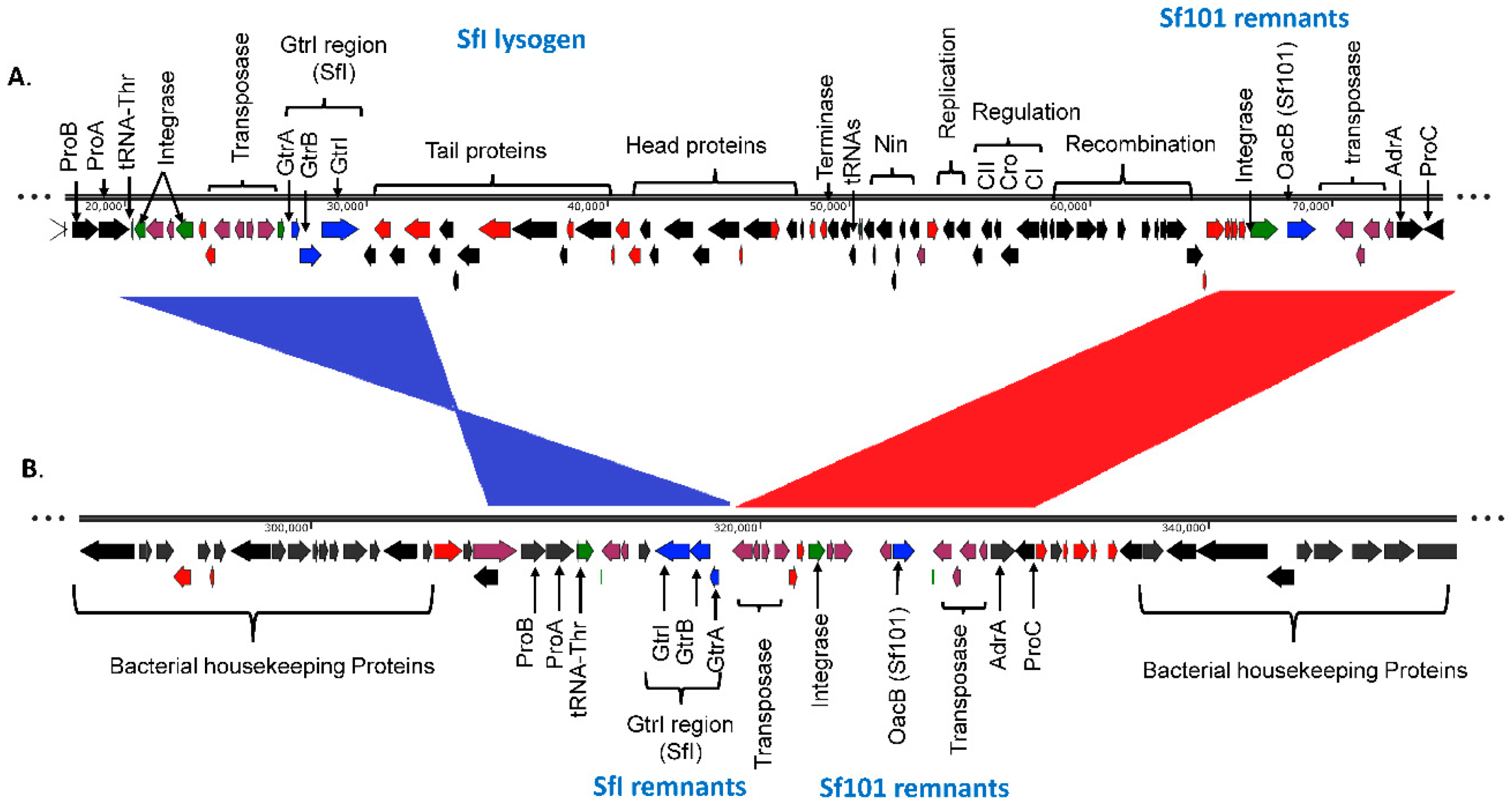
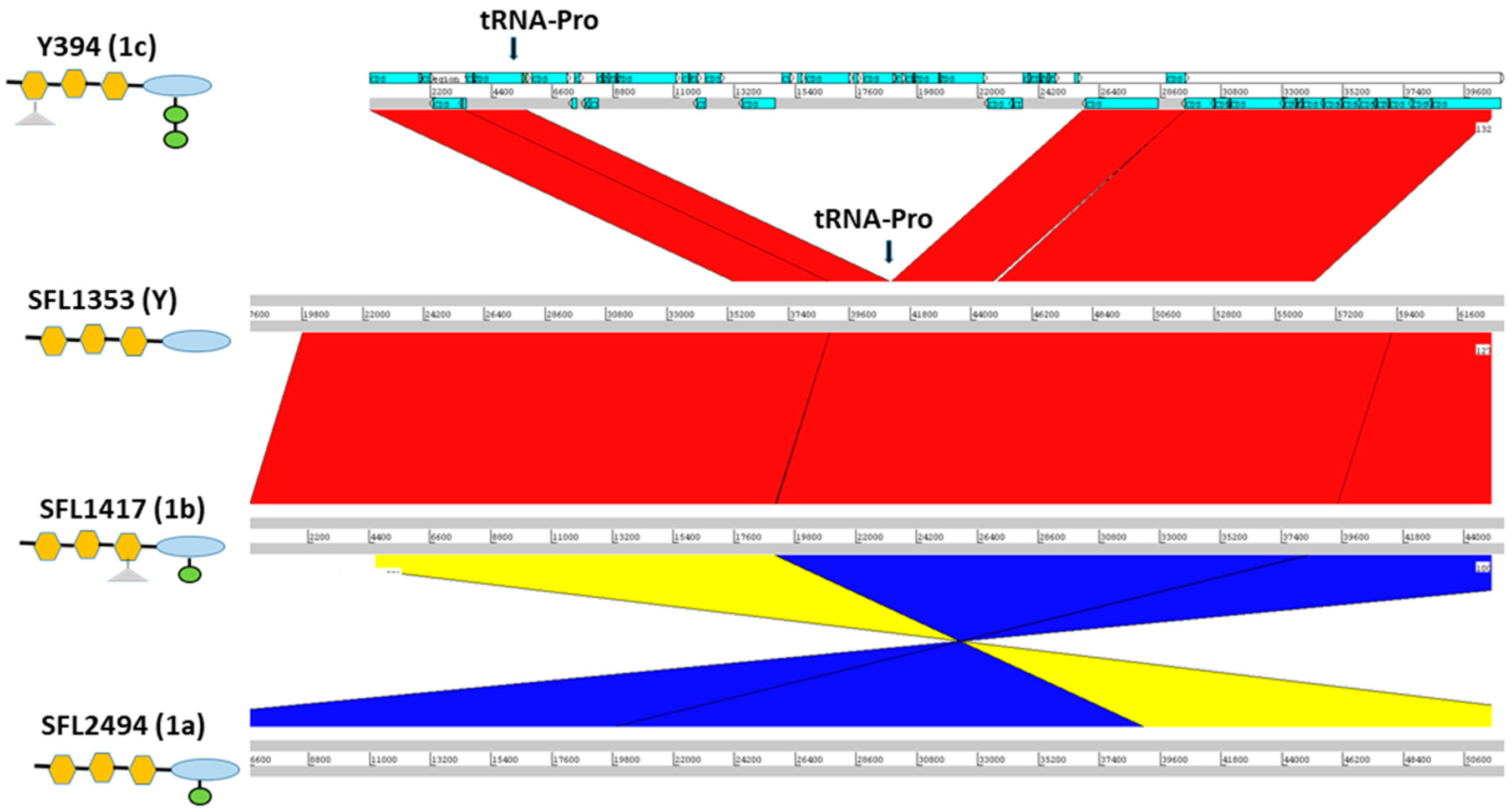
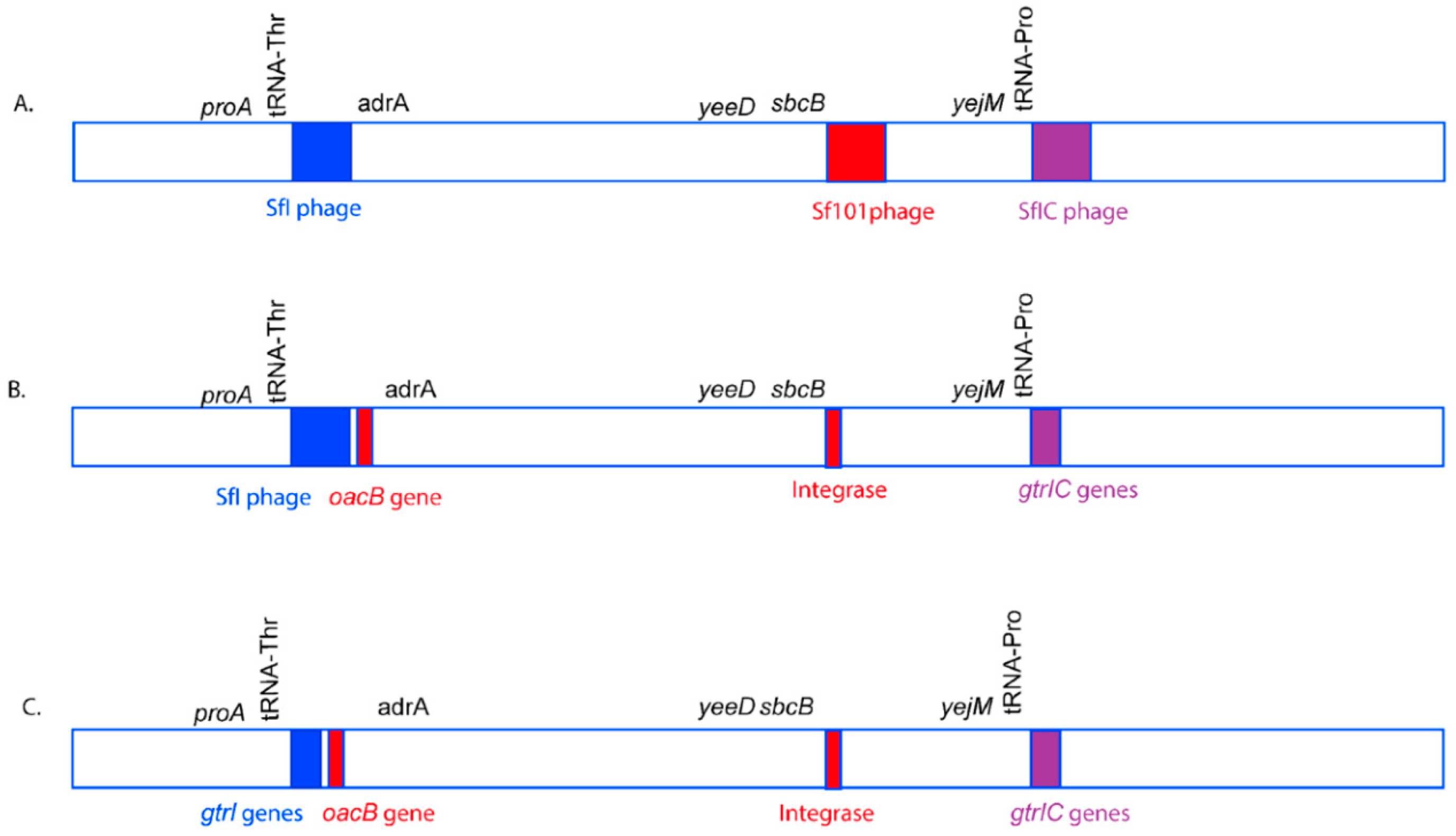
3.1. Integration of O-Antigen Modifying Bacteriophages in Serotype 1c Strains
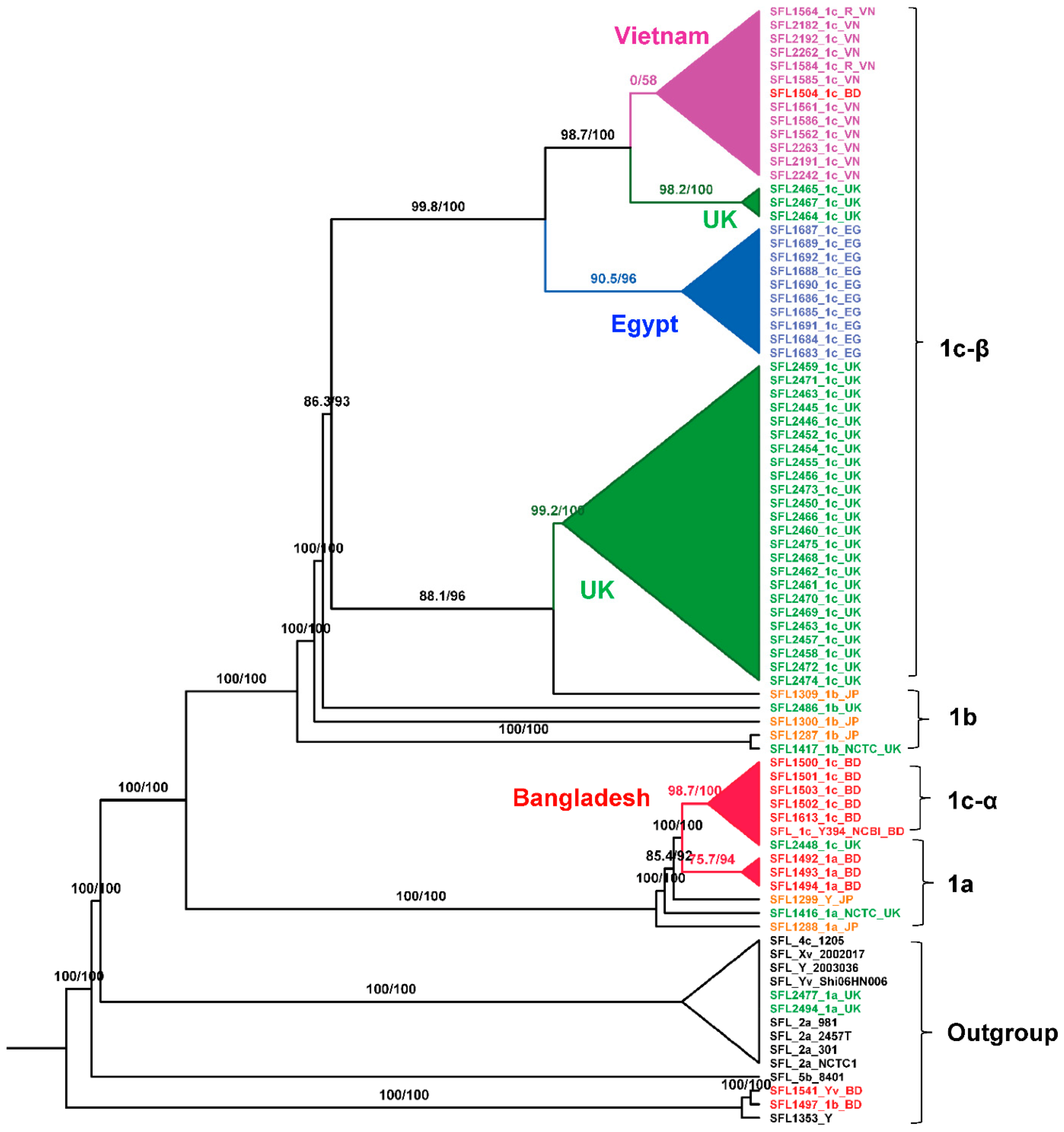
3.2. Pangenome and Phylogenetic Analysis
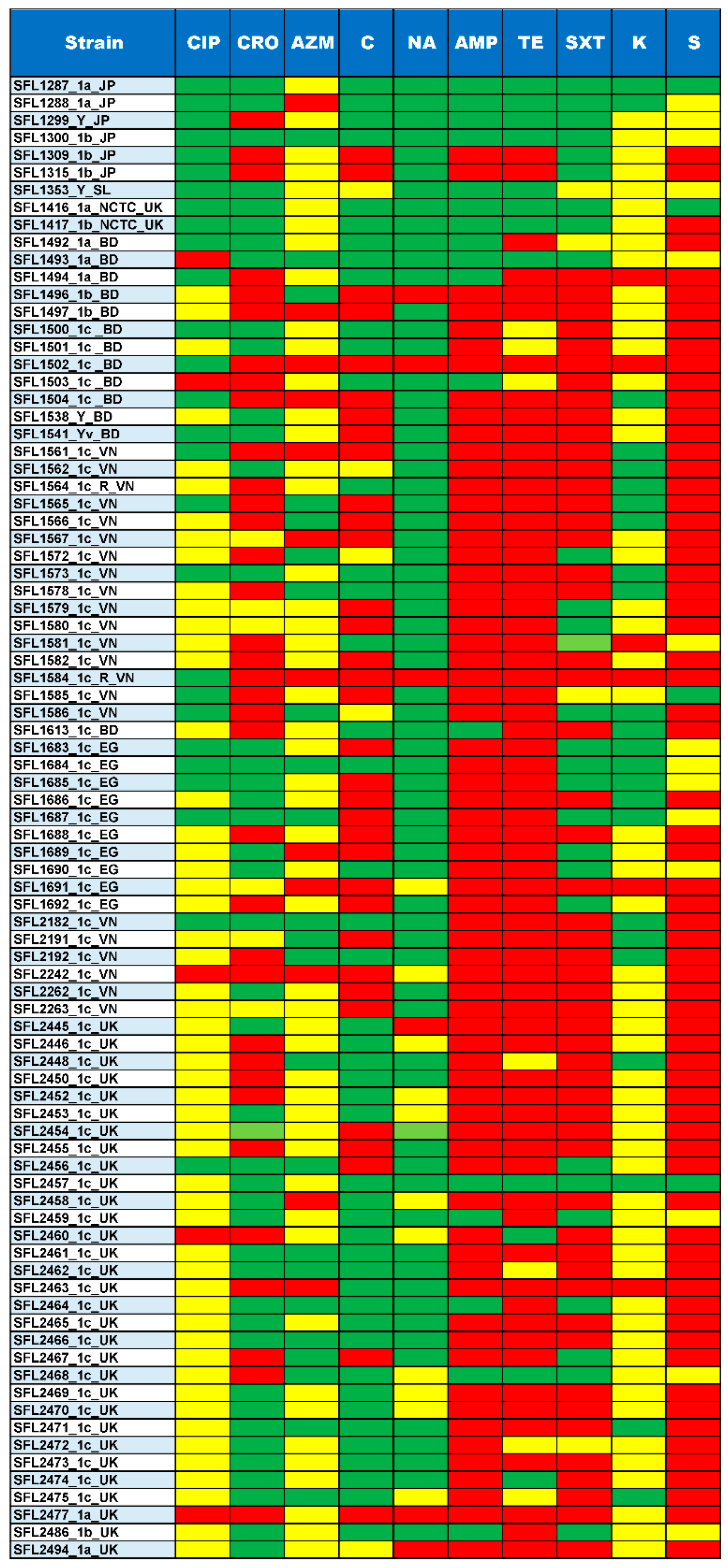
3.3. Antibiotic Resistance
4. Discussion
4.1. S. flexneri Serotype 1c Was Independently Evolved Twice from Serotype 1a and 1b Strains
4.2. Changing Landscape in Drug Resistance of S. flexneri
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Killackey, S.A.; Sorbara, M.T.; Girardin, S.E. Cellular aspects of shigella pathogenesis: Focus on the manipulation of host cell processes. Front. Cell Infect. Microbiol. 2016, 6, 38. [Google Scholar] [CrossRef] [PubMed]
- Maurelli, A.T. Shigella and enteroinvasive Escherichia coli: Paradigms for pathogen evolution and host–parasite interactions. In Escherichia Coli; Elsevier: Amsterdam, The Netherlands, 2013; pp. 215–245. [Google Scholar]
- Schwartz, B.S.; Harris, J.B.; Khan, A.I.; Larocque, R.C.; Sack, D.A.; Malek, M.A.; Faruque, A.S.; Qadri, F.; Calderwood, S.B.; Luby, S.P.; et al. Diarrheal epidemics in Dhaka, Bangladesh, during three consecutive floods: 1988, 1998, and 2004. Am. J. Trop. Med. Hyg. 2006, 74, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Kernéis, S.; Guerin, P.J.; von Seidlein, L.; Legros, D.; Grais, R.F. A look back at an ongoing problem: Shigella dysenteriae type 1 epidemics in refugee settings in Central Africa (1993–1995). PLoS ONE 2009, 4, e4494. [Google Scholar] [CrossRef] [PubMed]
- Lampel, K.A.; Formal, S.B.; Maurelli, A.T. A brief history of Shigella. EcoSal Plus 2018, 8. [Google Scholar] [CrossRef]
- Pires, S.M.; Fischer-Walker, C.L.; Lanata, C.F.; Devleesschauwer, B.; Hall, A.J.; Kirk, M.D.; Duarte, A.S.; Black, R.E.; Angulo, F.J. Aetiology-specific estimates of the global and regional incidence and mortality of diarrhoeal diseases commonly transmitted through food. PLoS ONE 2015, 10, e0142927. [Google Scholar] [CrossRef]
- Kotloff, K.L.; Blackwelder, W.C.; Nasrin, D.; Nataro, J.P.; Farag, T.H.; van Eijk, A.; Adegbola, R.A.; Alonso, P.L.; Breiman, R.F.; Faruque, A.S.; et al. The global enteric multicenter study (GEMS) of diarrheal disease in infants and young children in developing countries: Epidemiologic and clinical methods of the case/control study. Clin. Infect. Dis. 2012, 55 (Suppl. 4), S232–S245. [Google Scholar] [CrossRef]
- Bardhan, P.; Faruque, A.S.; Naheed, A.; Sack, D.A. Decrease in shigellosis-related deaths without shigella spp-specific interventions, Asia. Emerg. Infect. Dis. 2010, 16, 1718–1723. [Google Scholar] [CrossRef]
- Von Seidlein, L.; Kim, D.R.; Ali, M.; Lee, H.; Wang, X.; Thiem, V.D.; Canh, D.G.; Chaicumpa, W.; Agtini, M.D.; Hossain, A.; et al. A multicentre study of shigella diarrhoea in six Asian countries: Disease burden, clinical manifestations, and microbiology. PLoS Med. 2006, 3, e353. [Google Scholar] [CrossRef]
- Aragon, T.J.; Vugia, D.J.; Shallow, S.; Samuel, M.C.; Reingold, A.; Angulo, F.J.; Bradford, W.Z. Case-control study of shigellosis in San Francisco: The role of sexual transmission and HIV infection. Clin. Infect. Dis. 2007, 44, 327–334. [Google Scholar] [CrossRef]
- Kosek, M.; Yori, P.P.; Pan, W.K.; Olortegui, M.P.; Gilman, R.H.; Perez, J.; Chavez, C.B.; Sanchez, G.M.; Burga, R.; Hall, E. Epidemiology of highly endemic multiply antibiotic-resistant shigellosis in children in the Peruvian Amazon. Pediatrics 2008, 122, e541–e549. [Google Scholar] [CrossRef]
- Zaidi, M.B.; Estrada-Garcia, T.; Campos, F.D.; Chim, R.; Arjona, F.; Leon, M.; Michell, A.; Chaussabel, D. Incidence, clinical presentation, and antimicrobial resistance trends in Salmonella and Shigella infections from children in Yucatan, Mexico. Front. Microbiol. 2013, 4, 288. [Google Scholar] [CrossRef]
- Naik, D.G. Prevalence and antimicrobial susceptibility patterns of shigella species in Asmara, Eritrea, northeast Africa. J. Microbiol. Immunol. Infect. 2006, 39, 392–395. [Google Scholar]
- Kenne, L.; Lindberg, B.; Petersson, K.; Romanowska, E. Basic structure of the oligosaccharide repeating-unit of the Shigella flexneri O-antigens. Carbohydr. Res. 1977, 56, 363–370. [Google Scholar] [CrossRef]
- Sun, Q.; Lan, R.; Wang, J.; Xia, S.; Wang, Y.; Wang, Y.; Jin, D.; Yu, B.; Knirel, Y.A.; Xu, J. Identification and characterization of a novel shigella flexneri serotype Yv in China. PLoS ONE 2013, 8, e70238. [Google Scholar] [CrossRef] [PubMed]
- Parajuli, P.; Adamski, M.; Verma, N.K. Bacteriophages are the major drivers of shigella flexneri serotype 1c genome plasticity: A complete genome analysis. BMC Genom. 2017, 18, 722. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Klena, J.; Husain, T.; Monestersky, J.; Naguib, A.; Wasfy, M.O. Genetic characterization of antimicrobial resistance of shigella flexneri 1c isolates from patients in Egypt and Pakistan. Ann. Clin. Microbiol. Antimicrob. 2013, 12, 9. [Google Scholar] [CrossRef]
- El-Gendy, A.; El-Ghorab, N.; Lane, E.M.; Elyazeed, R.A.; Carlin, N.I.; Mitry, M.M.; Kay, B.A.; Savarino, S.J.; Peruski, L.F., Jr. Identification of Shigella flexneri subserotype 1c in rural Egypt. J. Clin. Microbiol. 1999, 37, 873–874. [Google Scholar] [CrossRef]
- Qiu, S.; Xu, X.; Wang, Y.; Yang, G.; Wang, Z.; Wang, H.; Zhang, L.; Liu, N.; Chen, C.; Liu, W.; et al. Emergence of resistance to fluoroquinolones and third-generation cephalosporins in Shigella flexneri subserotype 1c isolates from China. Clin. Microbiol. Infect. 2012, 18, E95–E98. [Google Scholar] [CrossRef] [PubMed]
- Stagg, R.M.; Cam, P.D.; Verma, N.K. Identification of newly recognized serotype 1c as the most prevalent shigella flexneri serotype in northern rural Vietnam. Epidemiol. Infect. 2008, 136, 1134–1140. [Google Scholar] [CrossRef]
- Wehler, T.; Carlin, N.I. Structural and immunochemical studies of the lipopolysaccharide from a new provisional serotype of Shigella flexneri. Eur. J. Biochem. 1988, 176, 471–476. [Google Scholar] [CrossRef]
- Guan, S.; Bastin, D.A.; Verma, N.K. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology 1999, 145, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Allison, G.E.; Verma, N.K. Serotype-converting bacteriophages and O-antigen modification in shigella flexneri. Trends Microbiol. 2000, 8, 17–23. [Google Scholar] [CrossRef]
- Stagg, R.M.; Tang, S.S.; Carlin, N.I.; Talukder, K.A.; Cam, P.D.; Verma, N.K. A novel glucosyltransferase involved in O-antigen modification of Shigella flexneri serotype 1c. J. Bacteriol. 2009, 191, 6612–6617. [Google Scholar] [CrossRef] [PubMed]
- Jakhetia, R.; Marri, A.; Stahle, J.; Widmalm, G.; Verma, N.K. Serotype-conversion in shigella flexneri: Identification of a novel bacteriophage, Sf101, from a serotype 7a strain. BMC Genom. 2014, 15, 742. [Google Scholar] [CrossRef] [PubMed]
- Pupo, G.M.; Lan, R.; Reeves, P.R. Multiple independent origins of Shigella clones of Escherichia coli and convergent evolution of many of their characteristics. Proc. Natl. Acad. Sci. USA 2000, 97, 10567–10572. [Google Scholar] [CrossRef]
- Yang, J.; Nie, H.; Chen, L.; Zhang, X.; Yang, F.; Xu, X.; Zhu, Y.; Yu, J.; Jin, Q. Revisiting the molecular evolutionary history of Shigella spp. J. Mol. Evol. 2007, 64, 71–79. [Google Scholar] [CrossRef]
- FastQC High Throughput Sequence QC Report. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 6 January 2020).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome project data processing s: The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef]
- Zerbino, D.R. Using the Velvet de novo assembler for short-read sequencing technologies. Curr. Protoc. Bioinform. 2010. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis comparison tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef]
- FigTree v1. 4. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 August 2020).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Cavalieri, S.J.; Harbeck, R.J.; McCarter, Y.S.; Ortez, J.H.; Rankin, I.D.; Sautter, R.L.; Sharp, S.E.; Spiegel, C.A. Manual of Antimicrobial Susceptibility Testing; American Society for Microbiology; Pan American Health Organization: Washington, DC, USA, 2005. [Google Scholar]
- Sun, Q.; Lan, R.; Wang, Y.; Wang, J.; Li, P.; Du, P.; Xu, J. Isolation and genomic characterization of SfI, a serotype-converting bacteriophage of Shigella flexneri. BMC Microbiol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2014, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Guidelines for the Control of Shigellosis, Including Epidemics Due to Shigella Dysenteriae Type 1; The World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Campbell, A. Prophage insertion sites. Res. Microbiol. 2003, 154, 277–282. [Google Scholar] [CrossRef]
- Hou, Y.-M. Transfer RNAs and pathogenicity islands. Trends Biochem. Sci. 1999, 24, 295–298. [Google Scholar] [CrossRef]
- Roberts, F.; Jennison, A.V.; Verma, N.K. The Shigella flexneri serotype Y vaccine candidate SFL124 originated from a serotype 2a background. FEMS Immunol. Med. Microbiol. 2005, 45, 285–289. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef]
- Brüssow, H.; Hendrix, R.W. Phage genomics: Small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef]
- Tang, S.S.; Carlin, N.I.; Talukder, K.A.; Cam, P.D.; Verma, N.K. Shigella flexneri serotype 1c derived from serotype 1a by acquisition of gtrIC gene cluster via a bacteriophage. BMC Microbiol. 2016, 16, 127. [Google Scholar] [CrossRef]
- Sack, D.A.; Lyke, C.; McLaughlin, C.; Suwanvanichkij, V.; World Health Organization (WHO). Antimicrobial Resistance in Shigellosis, Cholera and Campylobacteriosis; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- Bennish, M.L.; Salam, M.A.; Hossain, M.A.; Myaux, J.; Khan, E.H.; Chakraborty, J.; Henry, F.; Ronsmans, C. Antimicrobial resistance of shigella isolates in Bangladesh, 1983–1990: Increasing frequency of strains multiply resistant to ampicillin, trimethoprim-sulfamethoxazole, and nalidixic acid. Clin. Infect. Dis. 1992, 14, 1055–1060. [Google Scholar] [CrossRef]
- Bennish, M.L.; Salam, M.A.; Khan, W.A.; Khan, A.M. Treatment of shigellosis: III. Comparison of one-or two-dose ciprofloxacin with standard 5-day therapy: A randomized, blinded trial. Ann. Intern. Med. 1992, 117, 727–734. [Google Scholar] [CrossRef]
- Varsano, I.; Eidlitz-Marcus, T.; Nussinovitch, M.; Elian, I. Comparative efficacy of ceftriaxone and ampicillin for treatment of severe shigellosis in children. J. Pediatrics 1991, 118, 627–632. [Google Scholar] [CrossRef]
- Khatun, F.; Faruque, A.; Koeck, J.; Olliaro, P.; Millet, P.; Paris, N.; Malek, M.; Salam, M.; Luby, S. Changing species distribution and antimicrobial susceptibility pattern of Shigella over a 29-year period (1980–2008). Epidemiol. Infect. 2011, 139, 446–452. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parajuli, P.; Minh, B.Q.; Verma, N.K. Newly Emerged Serotype 1c of Shigella flexneri: Multiple Origins and Changing Drug Resistance Landscape. Genes 2020, 11, 1042. https://doi.org/10.3390/genes11091042
Parajuli P, Minh BQ, Verma NK. Newly Emerged Serotype 1c of Shigella flexneri: Multiple Origins and Changing Drug Resistance Landscape. Genes. 2020; 11(9):1042. https://doi.org/10.3390/genes11091042
Chicago/Turabian StyleParajuli, Pawan, Bui Quang Minh, and Naresh K. Verma. 2020. "Newly Emerged Serotype 1c of Shigella flexneri: Multiple Origins and Changing Drug Resistance Landscape" Genes 11, no. 9: 1042. https://doi.org/10.3390/genes11091042
APA StyleParajuli, P., Minh, B. Q., & Verma, N. K. (2020). Newly Emerged Serotype 1c of Shigella flexneri: Multiple Origins and Changing Drug Resistance Landscape. Genes, 11(9), 1042. https://doi.org/10.3390/genes11091042