Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features

 ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
2.2. Feature Extraction
2.3. Bi-Peptide-Based Evolutionary Feature Extraction Technique
- (i)
- The peptide sequence can be presented by P that is constituted as:P = R1 R2 R3 R4 … RL
- (ii)
- From Equation (5), we generate the transpose matrix as:with,where,
- (iii)
- The newly created matrix MTM is evaluated by the matrix M as well as the transpose of a matrix, which, in turn, is a 20 × 20 matrix (20L × L20 = 20 × 20 matrix) of 400 elements. The transpose matrix MT multiplies with the main M matrix. The resulting matrix is symmetric. Therefore, the upper or lower triangular plus the main diagonal will have all the information that is extracted in this matrix. The triangular matrix inhibits 210 features where the first 20 comes from the diagonal and the rest 190 ((400 − 20)/2) features are from either lower or upper triangle matrices (190 + 20 = 210 total), as shown below.
2.4. Handling Imbalanced Dataset
2.5. Classification Techniques
2.6. Performance Measurements
3. Results and Discussion
3.1. Building Our Model by Choosing the Most Effective Classifier
3.2. Comparison with State-of-the-Art Models
3.3. Web Server Implementation
4. Conclusions and Future Direction
Author Contributions
Funding
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Xu, Y.; Ding, J.; Wu, L.Y. iSulf-Cys: Prediction of S-sulfenylation sites in proteins with physicochemical properties of amino acids. PLoS ONE 2016, 11, e0154237. [Google Scholar] [CrossRef]
- Trost, B.; Maleki, F.; Kusalik, A.; Napper, S. DAPPLE 2: A Tool for the homology-based prediction of post-translational modification sites. J. Proteome. Res. 2016, 15, 2760–2767. [Google Scholar] [CrossRef]
- Trost, B.; Arsenault, R.; Griebel, P.; Napper, S.; Kusalik, A. DAPPLE: A pipeline for the homology-based prediction of phosphorylation sites. Bioinformatics 2013, 29, 1693–1695. [Google Scholar] [CrossRef][Green Version]
- Li, G.X.; Vogel, C.; Choi, H. PTMscape: An open source tool to predict genericpost-translational modifications and map modification crosstalk in proteindomains and biological processes. Mol. Omics. 2018, 14, 197–209. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, X.; Li, F.; Li, C.; Marquez-Lago, T.; Leier, A.; Akutsu, T.; Webb, G.; Dakang, X.; Smith, A.; et al. Large-scalecomparative assessment of computational predictors for lysine post-translationalmodification sites. Brief. Bioinf. 2019, 20, 2267–2290. [Google Scholar] [CrossRef]
- Xie, Z.; Dai, J.; Dai, L.; Tan, M.; Cheng, Z.; Wu, Y.; Boeke, J.D.; Zhao, Y. Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteom. 2012, 11, 100–107. [Google Scholar] [CrossRef]
- Kamynina, E.; Stover, P.J. The roles of SUMO in metabolic regulation. Adv. Exp. Med. Biol. 2017, 963, 143–168. [Google Scholar]
- Ju, Z.; He, J.J. Prediction of lysine propionylation sites using biased SVM and incorporating four different sequence features into Chou’s PseAAC. J. Mol. Gr. Modell. 2017, 76, 356–363. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Li, M.; Shyr, Y.; Xie, L.; Li, Y. Improved prediction of lysine acetylation by support vector machines. Protein Pept. Lett. 2009, 16, 977–983. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, M.; Xie, Z.; Dai, L.; Chen, Y.; Zhao, Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011, 7, 58–63. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. pSuc-Lys: Predict lysine succinylation sites in proteins with PseAAC and ensemble random forest approach. J. Theor. Biol. 2016, 394, 223–230. [Google Scholar] [CrossRef]
- Ahmad, M.W.; Arafat, M.E.; Taherzadeh, G.; Sharma, A.; Dipta, S.R.; Dehzangi, A.; Shatabda, S. Mal-light: Enhancing lysine malonylation sites prediction problem using evolutionary-based features. IEEE Access 2020, 8, 77888–77902. [Google Scholar] [CrossRef]
- Comb, D.G.; Sarkar, N.; Pinzino, C.J. The Methylation of lysine residues in protein. J. Biol. Chem. 1966, 241, 1857–1862. [Google Scholar]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Zhao, Y. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics. 2015, 14, 2308–2315. [Google Scholar] [CrossRef]
- Park, K.J.; Kanehisa, M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics 2003, 19, 1656–1663. [Google Scholar] [CrossRef]
- Xie, L.; Wang, G.; Yu, Z.; You, M.; Li, Q.; Huang, H.; Xie, J. Proteome-wide lysine glutarylation profiling of the Mycobacterium tuberculosis H37Rv. J. Proteome. Res. 2016, 15, 1379–1385. [Google Scholar] [CrossRef]
- Schmiesing, J.; Storch, S.; Dörfler, A.C.; Schweizer, M.; Makrypidi-Fraune, G.; Thelen, M.; Sylvester, M.; Gieselmann, V.; Meyer-Schwwsinger, C.; Koch-Nolte, F.; et al. Disease-linked glutarylation impairs function and interactions of mitochondrial proteins and contributes to mitochondrial heterogeneity. Cell Rep. 2018, 24, 2946–2956. [Google Scholar] [CrossRef]
- López, Y.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Tsunoda, T.; Sharma, A. SucStruct: Prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017, 527, 24–32. [Google Scholar] [CrossRef]
- López, Y.; Sharma, A.; Dehzangi, A.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T. Success: Evolutionary and structural properties of amino acids prove effective for succinylation site prediction. BMC Genom. 2018, 19, 923. [Google Scholar] [CrossRef]
- Ju, Z.; He, J.J. Prediction of lysine glutarylation sites by maximum relevance minimum redundancy feature selection. Anal. Biochem. 2018, 550, 1–7. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, Y.; Ding, J.; Li, C. iGlu-Lys: A predictor for lysine glutarylation through amino acid pair order features. IEEE Trans. NanoBiosci. 2018, 17, 394–401. [Google Scholar] [CrossRef]
- Huang, K.Y.; Kao, H.J.; Hsu, J.B.K.; Weng, S.L.; Lee, T.Y. Characterization and identification of lysine glutarylation based on intrinsic interdependence between positions in the substrate sites. BMC Bioinform. 2019, 19, 384. [Google Scholar] [CrossRef]
- AL-Barakati, H.J.; Saigo, H.; Newman, R.H. RF-GlutarySite: A random forest based predictor for glutarylation sites. Mol. Omics. 2019, 15, 189–204. [Google Scholar] [CrossRef]
- Chen, Z.; He, N.; Huang, Y.; Qin, W.T.; Liu, X.; Li, L. Integration of a deep learning classifier with a random forest approach for predicting malonylation sites. Genom. Proteom. Bioinf. 2018, 16, 451–459. [Google Scholar] [CrossRef]
- Wu, M.; Yang, Y.; Wang, H.; Xu, Y. A deep learning method to more accuratelyrecall known lysine acetylation sites. BMC Bioinform. 2019, 20, 49. [Google Scholar] [CrossRef]
- Chaudhari, M.; Thapa, N.; Roy, K.; Newman, R.; Saigo, H.; Dukka, B. DeepRMethylSite: A deep learning based approach for prediction of argininemethylation sites in proteins. Mol. Omics 2020. [Google Scholar] [CrossRef]
- Thapa, N.; Hiroto, S.; Roy, K.; Newman, R.H.; Dukka, K. RF-MaloSite andDL-MaloSite: Two independent computational methods based on random forest(RF) and deep learning (DL) to predict malonylation sites. Comput. Struct. Biotechnol. J. 2020, 18, 852–860. [Google Scholar]
- Xu, H.; Zhou, J.; Lin, S.; Deng, W.; Zhang, Y.; Xue, Y. PLMD: An updated data resource of protein lysine modifications. J. Gen. Genom. 2017, 44, 243–250. [Google Scholar] [CrossRef]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Paliwal, K.; Sharma, A.; Dehzangi, O.; Sattar., A. A combination of feature extraction methods with an ensemble of different classifiers for protein structural class prediction problem. IEEE/ACM Trans. Comput. Biol. Bioinform. 2013, 10, 564–575. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Michaelson, J.; Sattar, A.; Sharma, A. PSSM-Suc: Accurately predicting succinylation using position specific scoring matrix into bigram for feature extraction. J. Theor. Biol. 2017, 425, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Heffernan, R.; Sharma, A.; Lyons, J.; Paliwal, K.; Sattar, A. Gram-positive and gram-negative protein subcellular localization by incorporating evolutionary-based descriptors into chou’ s general PseAAC. J. Theor. Biol. 2015, 364, 284–294. [Google Scholar] [CrossRef]
- Chowdhury, S.Y.; Shatabda, S.; Dehzangi, A. iDNAprot-es: Identification of DNA-binding proteins using evolutionary and structural features. Sci. Rep. 2017, 7, 14938. [Google Scholar] [CrossRef]
- Ahmad, M.W.; Shovan, S.; Arafat, M.E.; Sifat, M.H.R.; Hasan, M.A.M.; Shatabda, S. Improved performance of lysine glutarylation PTM using peptide evolutionary features. In Proceedings of the 3rd International Conference on Electrical, Computer & Telecommunication Engineering (ICECTE) IEEE, RUET, Rajshahi, Bangladesh, 26–28 December 2019. [Google Scholar]
- Sharma, A.; Lyons, J.; Dehzangi, A.; Paliwal, K.K. A feature extraction technique using bi-gram probabilities of position specific scoring matrix for protein fold recognition. J. Theor. Biol. 2013, 320, 41–46. [Google Scholar] [CrossRef]
- Ahmed, M.W.; Arafat, M.E.; Shovan, S.M.; Uddin, M.; Osama, O.F.; Shatabda, S. Enhanced prediction of lysine propionylation sites using Bi-peptide evolutionary features resolving data imbalance. In Proceedings of the IEEE Region 10 Symposium (TENSYMP 2020), Dhaka, Bangladesh, 5–7 April 2020. [Google Scholar]
- Dehzangi, A.; López, Y.; Lal, S.P.; Taherzadeh, G.; Sattar, A.; Tsunoda, T.; Sharma, A. Improving succinylation prediction accuracy by incorporating the secondary structure via helix, strand and coil, and evolutionary information from profile bigrams. PLoS ONE 2018, 13, E0191900. [Google Scholar] [CrossRef]
- Shatabda, S.; Saha, S.; Sharma, A.; Dehzangi, A. iPHLoc-ES: Identification of bacteriophage protein locations using evolutionary and structural features. J. Theor. Biol. 2017, 435, 229–237. [Google Scholar] [CrossRef]
- Schaffer, A. Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucl. Acids Res. 2001, 29, 2994–3005. [Google Scholar] [CrossRef]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iSuc-PseOpt: Identifying lysine succinylation sites in proteins by incorporating sequence-coupling effects into pseudo components and optimizing imbalanced training dataset. Anal. Biochem. 2016, 497, 48–56. [Google Scholar] [CrossRef]
- Chandra, A.; Sharma, A.; Dehzangi, A.; Ranganathan, S.; Jokhan, A.; Chou, K.C.; Tsunoda, T. PhoglyStruct: Prediction of phosphoglycerylated lysine residues using structural properties of amino acids. Sci. Rep. 2018, 8, 17923. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Liu, Z.; Xiao, X.; Liu, B.; Chou, K.C. iPPBS-Opt: A sequence-Based ensemble classifier for identifying protein-protein binding sites by optimizing imbalanced training datasets. Molecules 2016, 21, 95. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Zuo, Y. S-SulfPred: A sensitive predictor to capture S-sulfenylation sites based on a resampling one-sided selection undersampling-synthetic minority oversampling technique. J. Theor. Biol. 2017, 422, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Hasan, M.M.; Khatun, M.S.; Kurata, H. A comprehensive review of in silico analysis for protein S-sulfenylation sites. Protein Pept. Lett. 2018, 25, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Li, J.; Gu, L.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.D. Identifying the characteristics of the hypusination sites using SMOTE and SVM algorithmwith feature selection. Curr. Proteom. 2018, 15, 111–118. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, R.; Wang, J.; Leier, A.; Marquez-Lago, T.T.; Akutsu, T.; Song, J. Computational analysis and prediction of lysine malonylation sites by exploiting informative features in an integrative machine-learning framework. Brief. Bioinform. 2018, 5, 2185–2199. [Google Scholar] [CrossRef]
- Iqbal, S.; Hoque, M.T. PBRpredict-Suite: A suite of models to predict peptide-recognition domain residues from protein sequence. Bioinformatics 2018, 34, 3289–3299. [Google Scholar] [CrossRef]
- Zahiri, J.; Mohammad-Noori, M.; Ebrahimpour, R.; Saadat, S.; Bozorgmehr, J.H.; Goldberg, T.; Masoudi-Nejad, A. LocFuse: Human protein–protein interaction prediction via classifier fusion using protein localization information. Genomics 2014, 104, 496–503. [Google Scholar] [CrossRef]
- Ismail, H.D.; Newman, R.H. RF-Hydroxysite: A random forest based predictor for hydroxylation sites. Mol. BioSyst. 2016, 12, 2427–2435. [Google Scholar] [CrossRef]
- Jiao, Y.; Du, P. Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 2016, 4, 320–330. [Google Scholar] [CrossRef]
- Chen, C.Y.; Tang, S.L.; Chou, S.C.T. Taxonomy based performance metrics for evaluating taxonomic assignment methods. BMC Bioinform. 2019, 20, 310. [Google Scholar] [CrossRef] [PubMed]
- Dehzangi, A.; Paliwal, K.; Lyons, J.; Sharma, A.; Sattar, A. Enhancing protein fold prediction accuracy using evolutionary and structural features. In Proceedings of the IAPR International Conference on Pattern Recognition Bioinformatics; 17–20 June 2013; pp. 196–207. [CrossRef]
- Chen, Z.; Zhao, P.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Wang, Y.; Webb, G.I.; Smith, A.I.; Daly, R.J.; Chou, K.C.; et al. iFeature: A python package and web server for features extraction and selection from protein and peptide sequences. Bioinformatics 2018, 34, 2499–2502. [Google Scholar] [CrossRef] [PubMed]
- Liu, B. BioSeq-Analysis: A platform for DNA, RNA, and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2019, 20, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Gao, X.; Zhang, H. BioSeq-Analysis2.0: An updated platform for analyzing DNA, RNA, and protein sequences at sequence level and residue level based on machine learning approaches. Nucl. Acids Res. 2019, 47, e127. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Vans, E.; Shigemizu, D.; Boroevich, K.A.; Tsunoda, T. DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
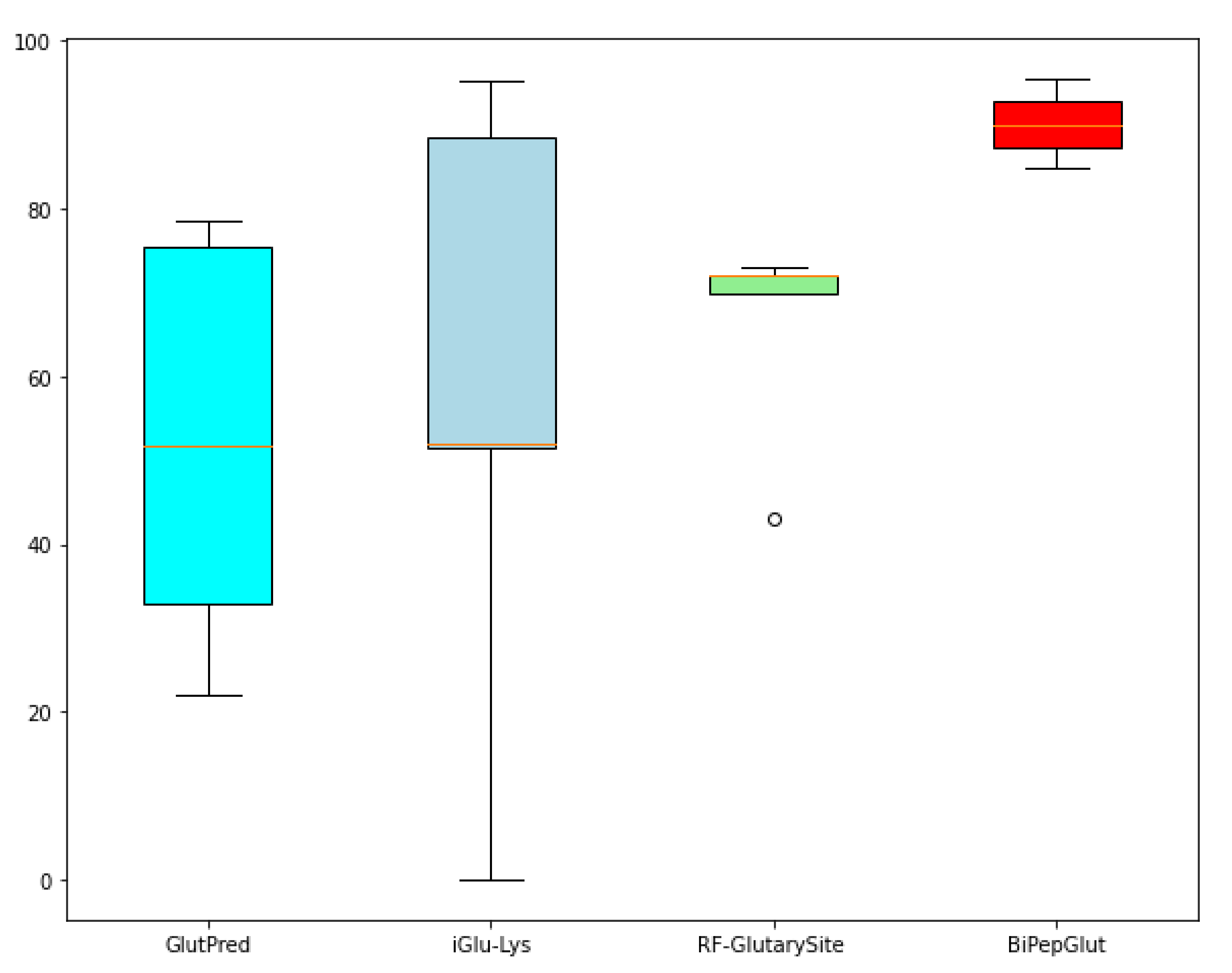{kind=link}
{kind=link}
| Model | ACC (%) | SN (%) | SP (%) | MCC | F1-Score |
|---|---|---|---|---|---|
| RF | 80.2% | 63.4% | 96.9% | 0.64 | 0.76 |
| XGBoost | 79.7% | 67.8% | 91.5% | 0.61 | 0.76 |
| LightGBM | 82.9% | 74.2% | 91.5% | 0.67 | 0.81 |
| AdaBoost | 79.2% | 74.7% | 83.8% | 0.59 | 0.78 |
| ET classifier | 81.5% | 70.0% | 92.9% | 0.64 | 0.79 |
| MLP | 78.7% | 75.4% | 82.0% | 0.58 | 0.78 |
| Classifier Model | ACC (%) | SN (%) | SP (%) | MCC | F1-Score |
|---|---|---|---|---|---|
| RF | 79.6% | 41.3% | 98.9% | 0.54 | 0.58 |
| XGBoost | 80.3% | 45.7% | 97.8% | 0.55 | 0.61 |
| LightGBM | 91.2% | 78.3% | 97.8% | 0.80 | 0.86 |
| AdaBoost | 85.4% | 76.1% | 90.1% | 0.67 | 0.78 |
| ET Classifier | 92.0% | 84.8% | 95.6% | 0.82 | 0.88 |
| MLP | 84.7% | 76.1% | 88.0% | 0.64 | 0.76 |
| Predictor Tool | ACC (%) | SN (%) | SP (%) | MCC | F1-Score |
|---|---|---|---|---|---|
| GlutPred [22] | 74.9% | 64.8% | 76.6% | 0.32 | 0.43 |
| iGlu-Lys [23] | 88.4% | 50.4% | 95.2% | 0.51 | - |
| RF-GlutarySite [25] | 75.0% | 81.0% | 68.0% | 0.50 | 0.73 |
| BiPepGlut | 81.5% | 70.0% | 92.9% | 0.64 | 0.79 |
| Predictor Tool | ACC (%) | SN (%) | SP (%) | MCC | F1-Score |
|---|---|---|---|---|---|
| GlutPred [22] | 75.4% | 51.8% | 78.5% | 0.22 | 0.33 |
| iGlu-Lys [23] | 88.5% | 51.4% | 95.3% | 0.52 | - |
| RF-GlutarySite [25] | 72.0% | 73.0% | 70.0% | 0.43 | 0.72 |
| BiPepGlut | 92.0% | 84.8% | 95.6% | 0.82 | 0.88 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arafat, M.E.; Ahmad, M.W.; Shovan, S.M.; Dehzangi, A.; Dipta, S.R.; Hasan, M.A.M.; Taherzadeh, G.; Shatabda, S.; Sharma, A. Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features. Genes 2020, 11, 1023. https://doi.org/10.3390/genes11091023
Arafat ME, Ahmad MW, Shovan SM, Dehzangi A, Dipta SR, Hasan MAM, Taherzadeh G, Shatabda S, Sharma A. Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features. Genes. 2020; 11(9):1023. https://doi.org/10.3390/genes11091023
Chicago/Turabian StyleArafat, Md. Easin, Md. Wakil Ahmad, S.M. Shovan, Abdollah Dehzangi, Shubhashis Roy Dipta, Md. Al Mehedi Hasan, Ghazaleh Taherzadeh, Swakkhar Shatabda, and Alok Sharma. 2020. "Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features" Genes 11, no. 9: 1023. https://doi.org/10.3390/genes11091023
APA StyleArafat, M. E., Ahmad, M. W., Shovan, S. M., Dehzangi, A., Dipta, S. R., Hasan, M. A. M., Taherzadeh, G., Shatabda, S., & Sharma, A. (2020). Accurately Predicting Glutarylation Sites Using Sequential Bi-Peptide-Based Evolutionary Features. Genes, 11(9), 1023. https://doi.org/10.3390/genes11091023