Identification of circRNA-lncRNA-miRNA-mRNA Competitive Endogenous RNA Network as Novel Prognostic Markers for Acute Myeloid Leukemia

 , ,
, ,
Abstract
1. Introduction
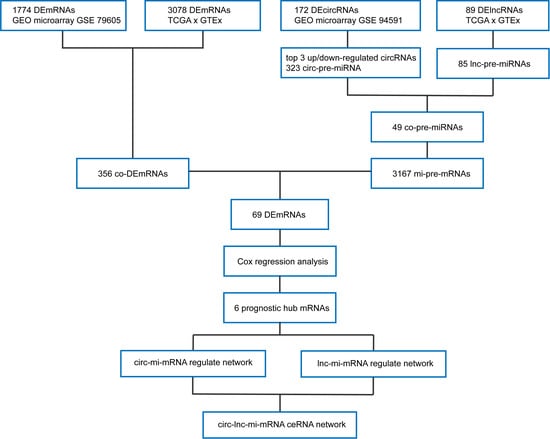
2. Materials and Methods
2.1. Transcriptome Data and Clinical Information
2.2. Identification of DERNAs
2.3. Gene Function Annotation
2.4. Prediction of Targeting Relationship
2.5. Survival Prediction Model Identifies Hub mRNAs
2.6. ceRNA Network Establishment
2.7. Statistical Analysis
3. Results
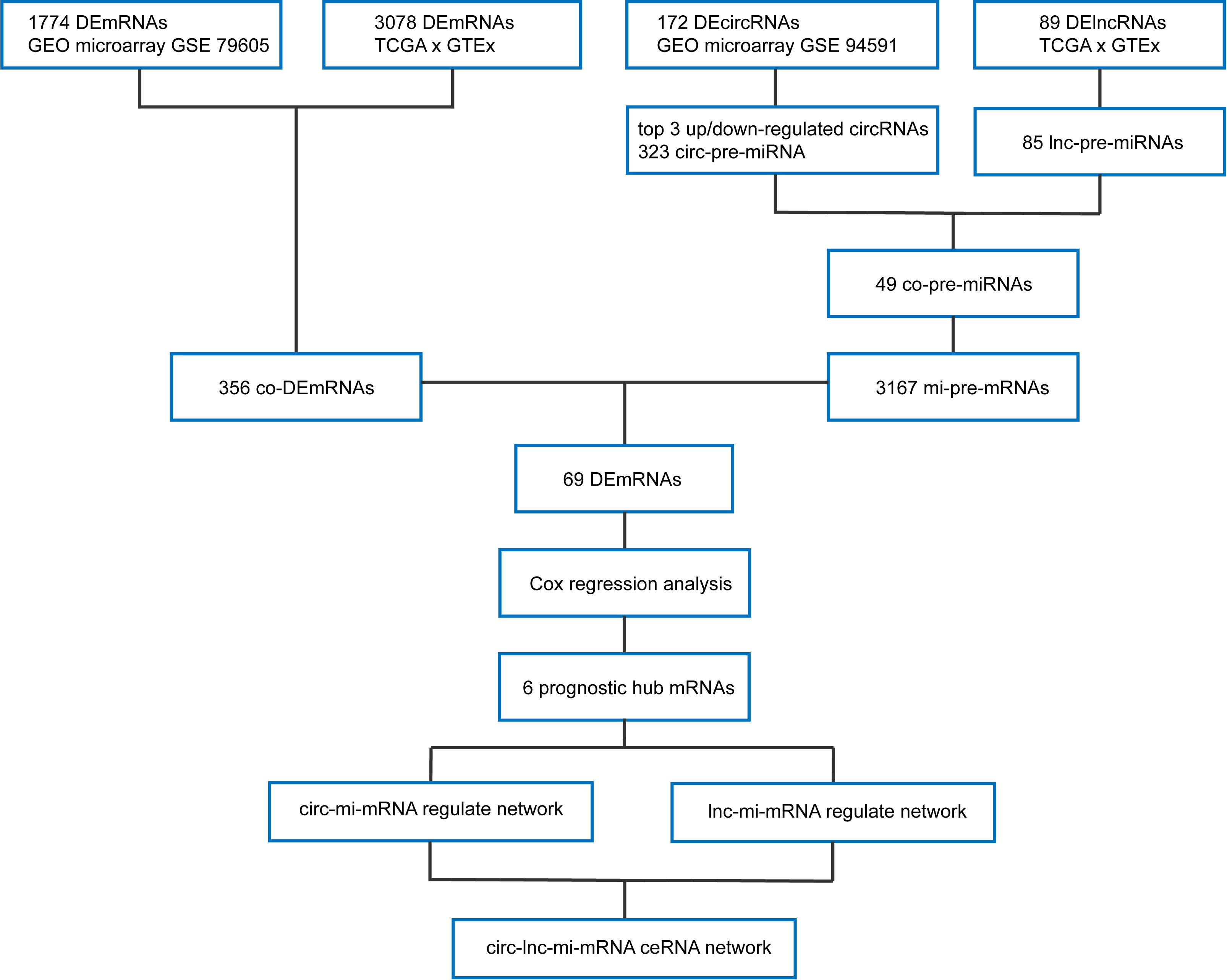
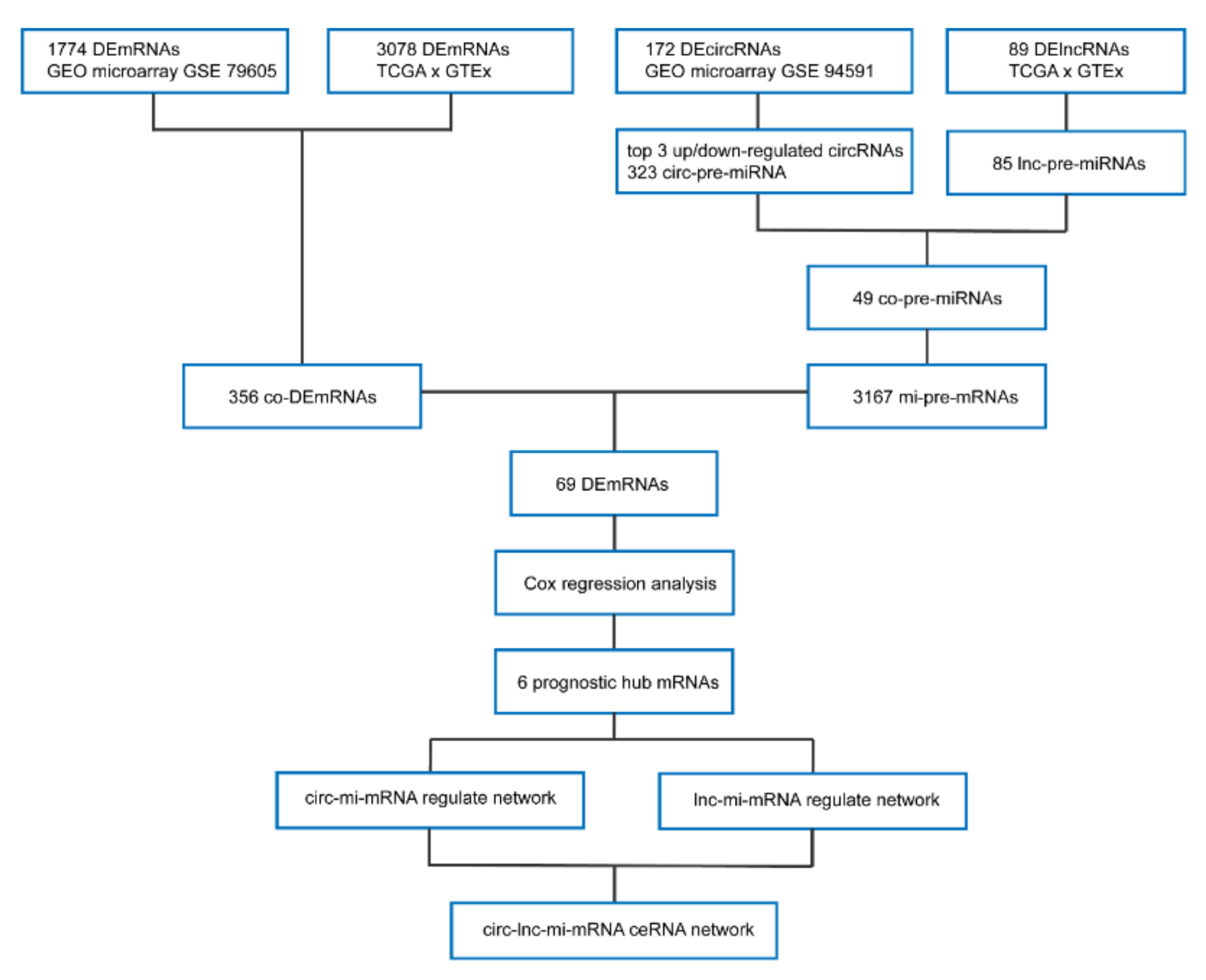
3.1. Identification of DEmRNAs between AML and Normal Samples
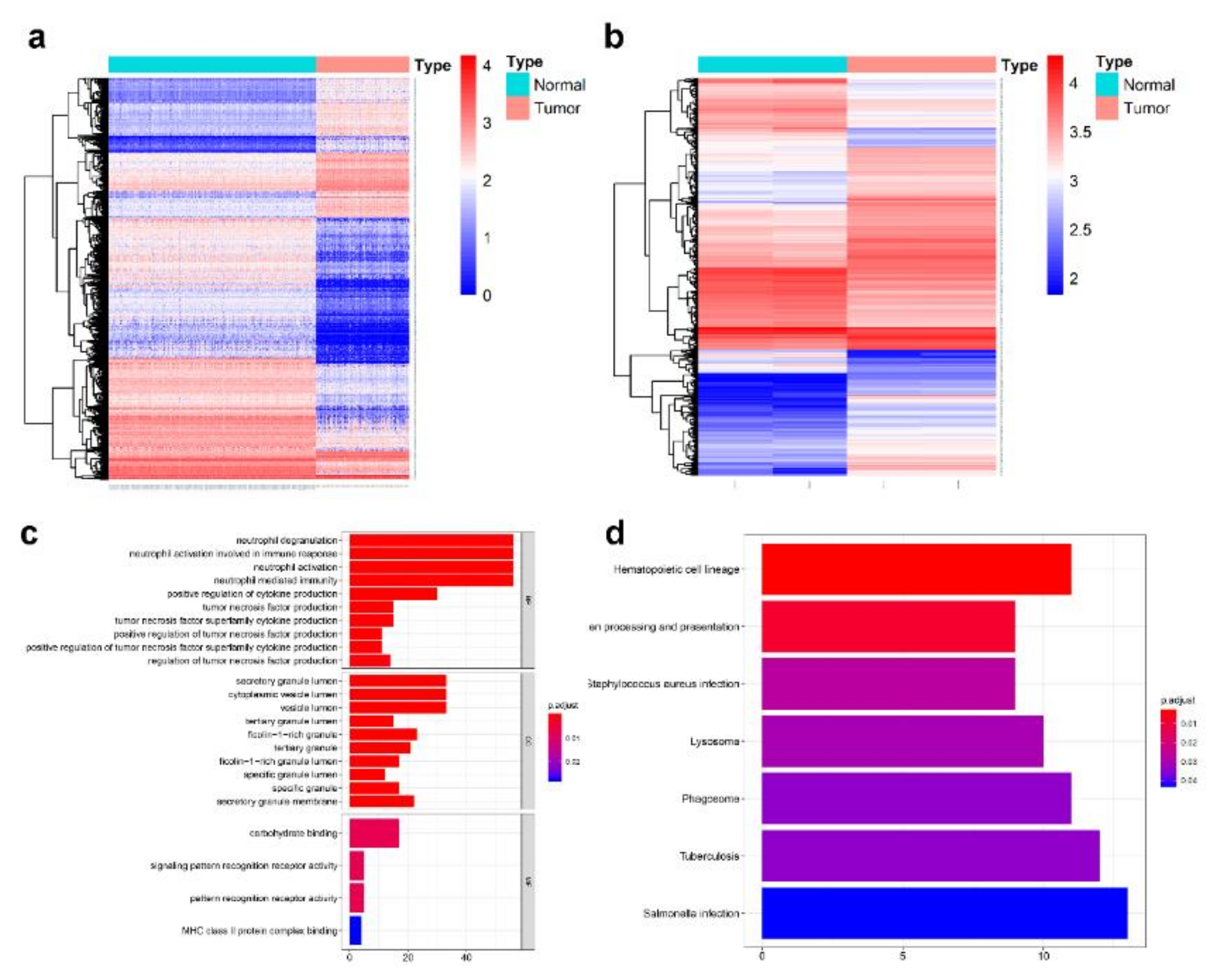
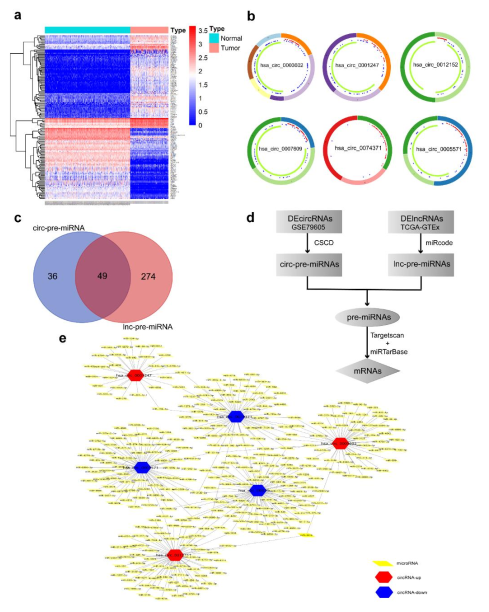
3.2. Identification of DelncRNAs and DEcircRNAs between AML and Normal Samples
3.3. Prediction and Identification miRNAs Targeted by Both lncRNA and circRNA
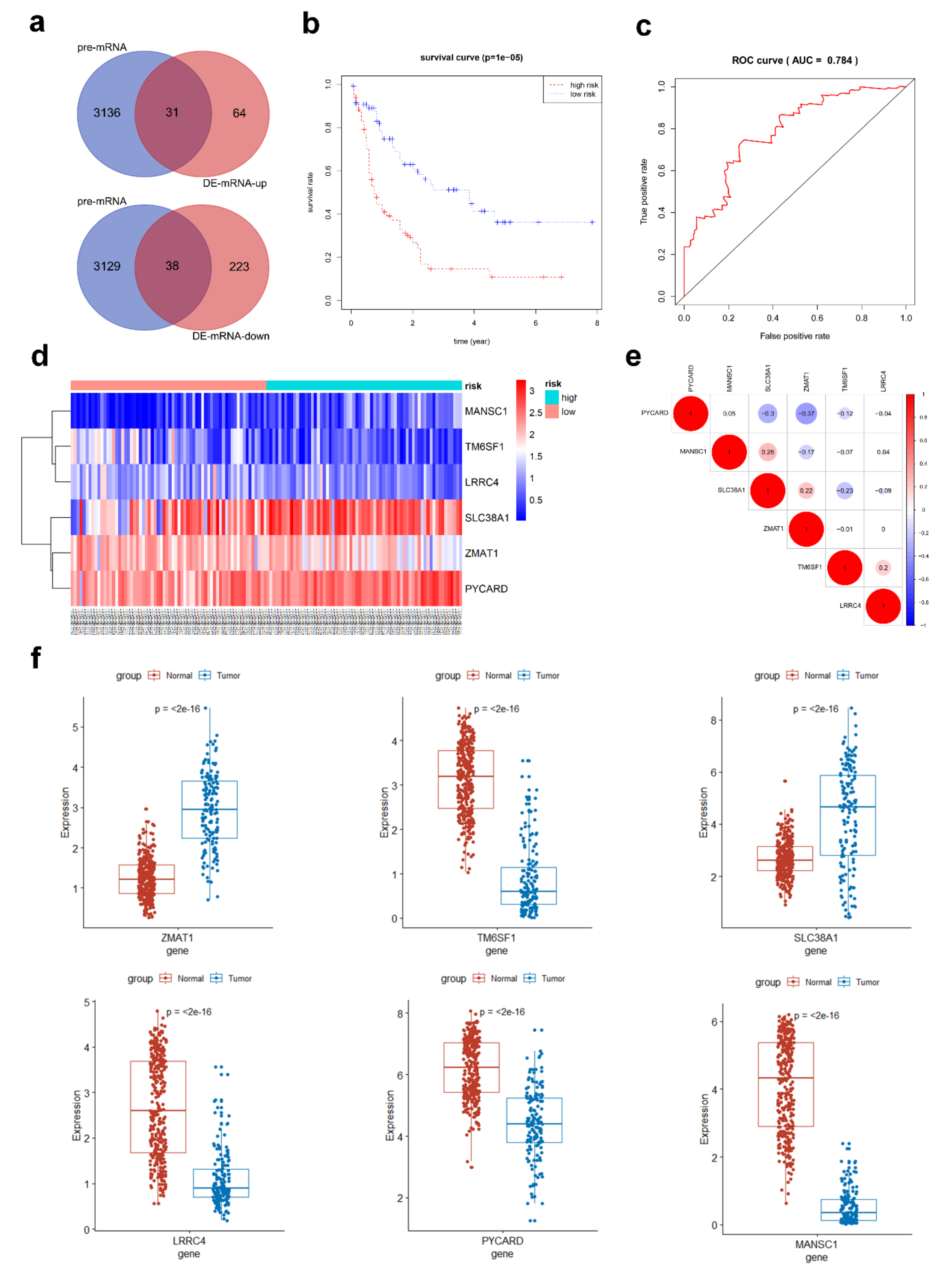
3.4. Prediction of miRNA-mRNA Targeting Relationship and Screening for mRNAs Highly Related to AML
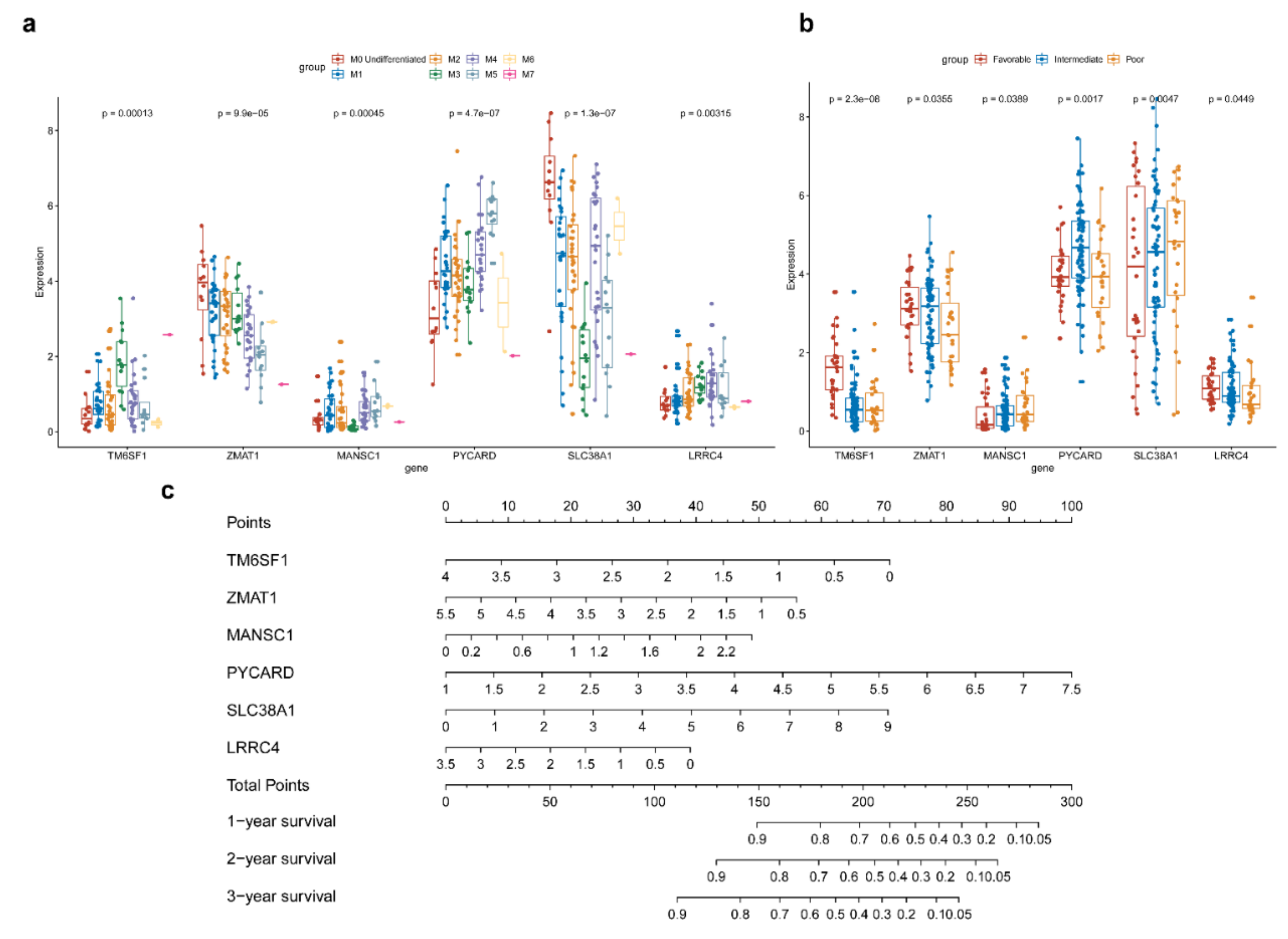
3.5. Hub AML Prognostic mRNAs Screened by Cox Regression
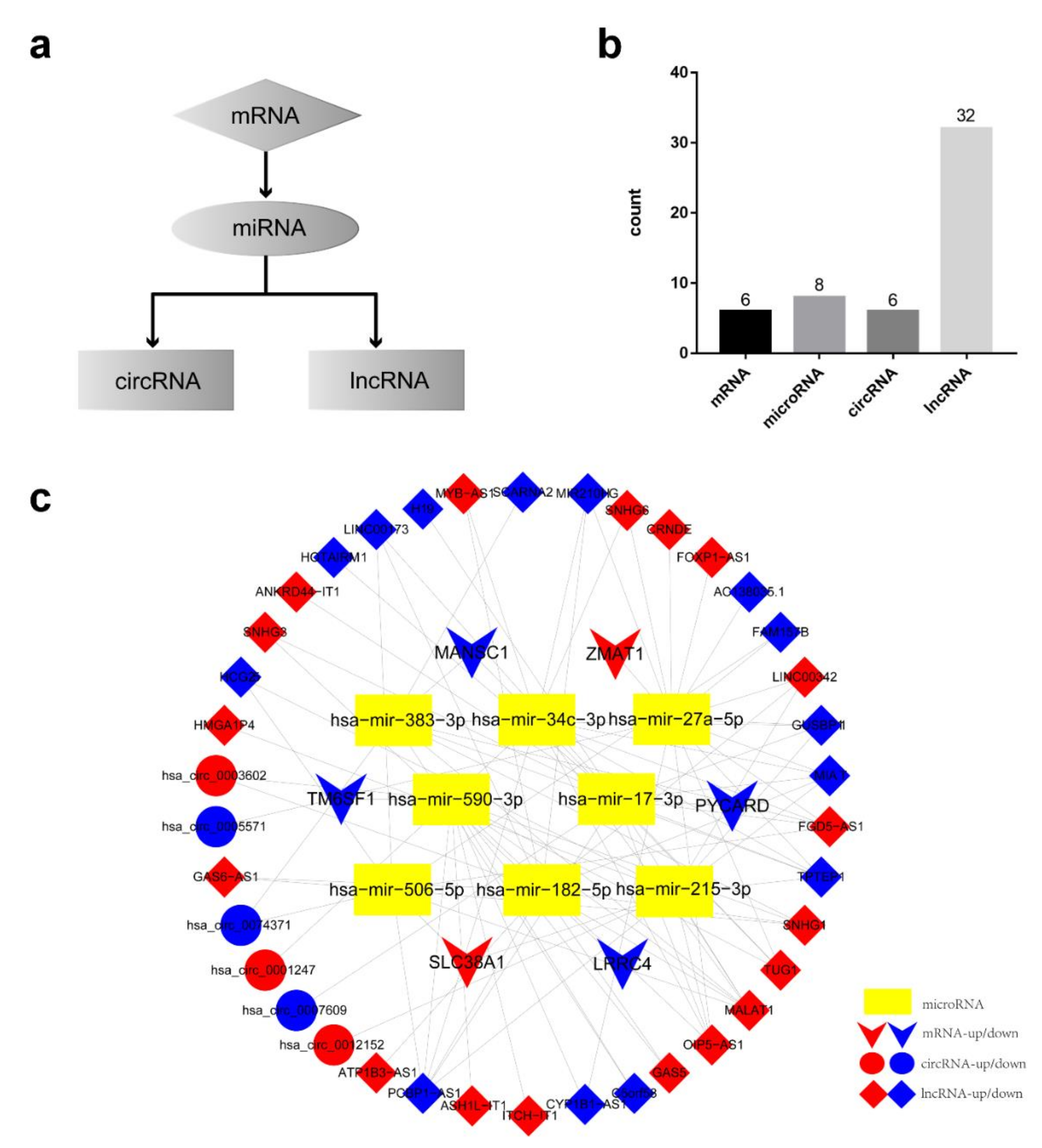
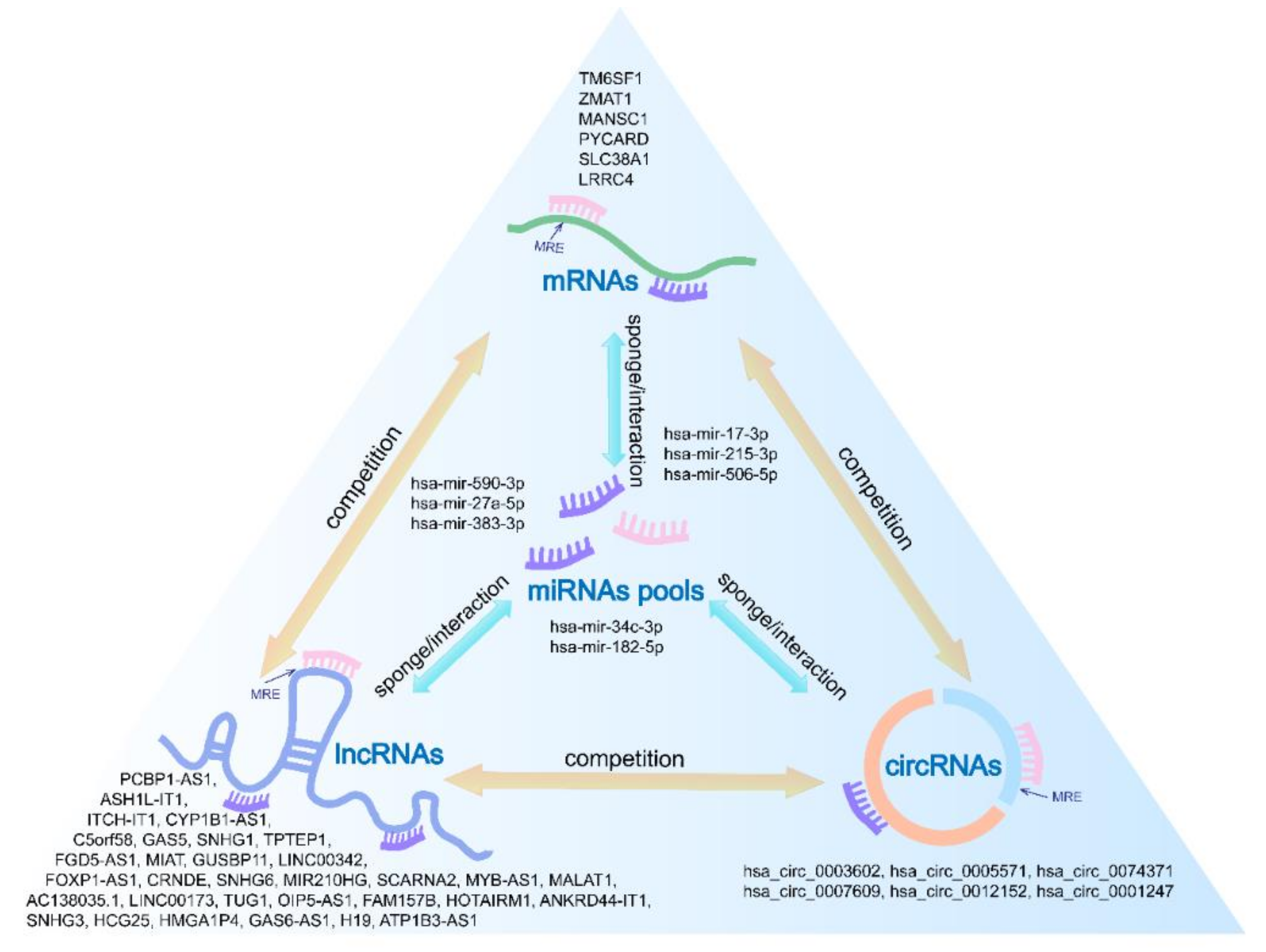
3.6. Establishment of AML Prognostic circRNA-lncRNA-miRNA-mRNA ceRNA Network
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Juliusson, G.; Lazarevic, V.; Horstedt, A.S.; Hagberg, O.; Hoglund, M. Swedish Acute Leukemia Registry, G. Acute myeloid leukemia in the real world: Why population-based registries are needed. Blood 2012, 119, 3890–3899. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Stolzel, F.; Rollig, C.; Radke, J.; Mohr, B.; Platzbecker, U.; Bornhauser, M.; Paulus, T.; Ehninger, G.; Zophel, K.; Schaich, M. (1)(8)f-fdg-pet/ct for detection of extramedullary acute myeloid leukemia. Haematologica 2011, 96, 1552–1556. [Google Scholar] [CrossRef]
- Burnett, A.; Wetzler, M.; Lowenberg, B. Therapeutic advances in acute myeloid leukemia. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 487–494. [Google Scholar] [CrossRef]
- Cornelissen, J.J.; Blaise, D. Hematopoietic stem cell transplantation for patients with aml in first complete remission. Blood 2016, 127, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microrna sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.; Chen, L.L. The biogenesis, functions, and challenges of circular rnas. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding rnas. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Tay, Y.; Rinn, J.; Pandolfi, P.P. The multilayered complexity of cerna crosstalk and competition. Nature 2014, 505, 344–352. [Google Scholar] [CrossRef]
- Cheng, D.L.; Xiang, Y.Y.; Ji, L.J.; Lu, X.J. Competing endogenous RNA interplay in cancer: Mechanism, methodology, and perspectives. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2015, 36, 479–488. [Google Scholar] [CrossRef]
- Wu, D.M.; Wen, X.; Han, X.R.; Wang, S.; Wang, Y.J.; Shen, M.; Fan, S.H.; Zhang, Z.F.; Shan, Q.; Li, M.Q.; et al. Role of circular RNA dleu2 in human acute myeloid leukemia. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Jamal, M.; Song, T.; Chen, B.; Faisal, M.; Hong, Z.; Xie, T.; Wu, Y.; Pan, S.; Yin, Q.; Shao, L.; et al. Recent progress on circular RNA research in acute myeloid leukemia. Front. Oncol. 2019, 9, 1108. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.M.; Ma, J.; Fang, W.B. Identification of non-coding RNA regulatory networks in pediatric acute myeloid leukemia reveals circ-0004136 could promote cell proliferation by sponging mir-142. Eur. Rev. Med Pharmacol. Sci. 2019, 23, 9251–9258. [Google Scholar] [PubMed]
- Lei, B.; He, A.; Chen, Y.; Cao, X.; Zhang, P.; Liu, J.; Ma, X.; Qian, L.; Zhang, W. Long non-coding RNA rpph1 promotes the proliferation, invasion and migration of human acute myeloid leukemia cells through down-regulating mir-330-5p expression. EXCLI J. 2019, 18, 824–837. [Google Scholar] [PubMed]
- Liu, Y.; Gao, X.; Tian, X. High expression of long intergenic non-coding RNA linc00662 contributes to malignant growth of acute myeloid leukemia cells by upregulating rock1 via sponging microrna-340-5p. Eur. J. Pharmacol. 2019, 859, 172535. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Zhou, H.S.; Tu, X.X.; He, Y.; Liu, Q.F.; Liu, Q.; Long, Z.J. Prediction of competing endogenous RNA coexpression network as prognostic markers in aml. Aging 2019, 11, 3333–3347. [Google Scholar] [CrossRef]
- Lv, C.; Sun, L.; Guo, Z.; Li, H.; Kong, D.; Xu, B.; Lin, L.; Liu, T.; Guo, D.; Zhou, J.; et al. Circular RNA regulatory network reveals cell-cell crosstalk in acute myeloid leukemia extramedullary infiltration. J. Transl. Med. 2018, 16, 361. [Google Scholar] [CrossRef]
- Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J.; Laird, P.W.; Baty, J.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar]
- Consortium, G.T. The genotype-tissue expression (gtex) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar]
- Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a new biomarker for acute myeloid leukemia via circular RNA profile and bioinformatics analysis. Int. J. Mol. Sci. 2017, 18, E597. [Google Scholar] [CrossRef]
- Gargouri, M.; Park, J.J.; Holguin, F.O.; Kim, M.J.; Wang, H.; Deshpande, R.R.; Shachar-Hill, Y.; Hicks, L.M.; Gang, D.R. Identification of regulatory network hubs that control lipid metabolism in chlamydomonas reinhardtii. J. Exp. Bot. 2015, 66, 4551–4566. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Peng, S.; Wang, J.; Wei, C.; Ye, Z.; Wang, Y.; Wang, M.; Xu, H.; Jiang, S.; Sun, D.; et al. Identification and characterization of circrnas as competing endogenous rnas for mirna-mrna in colorectal cancer. PeerJ 2019, 7, e7602. [Google Scholar] [CrossRef] [PubMed]
- Glazar, P.; Papavasileiou, P.; Rajewsky, N. Circbase: A database for circular rnas. RNA 2014, 20, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Feng, J.; Chen, K.; Ma, Y.; Gong, J.; Cai, F.; Jin, Y.; Gao, Y.; Xia, L.; Chang, H.; et al. Cscd: A database for cancer-specific circular rnas. Nucleic Acids Res. 2018, 46, D925–D929. [Google Scholar] [CrossRef] [PubMed]
- Jeggari, A.; Marks, D.S.; Larsson, E. Mircode: A map of putative microrna target sites in the long non-coding transcriptome. Bioinformatics 2012, 28, 2062–2063. [Google Scholar] [CrossRef] [PubMed]
- Fromm, B.; Billipp, T.; Peck, L.E.; Johansen, M.; Tarver, J.E.; King, B.L.; Newcomb, J.M.; Sempere, L.F.; Flatmark, K.; Hovig, E.; et al. A uniform system for the annotation of vertebrate microrna genes and the evolution of the human micrornaome. Annu. Rev. Genet. 2015, 49, 213–242. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.D.; Lin, F.M.; Wu, W.Y.; Liang, C.; Huang, W.C.; Chan, W.L.; Tsai, W.T.; Chen, G.Z.; Lee, C.J.; Chiu, C.M.; et al. Mirtarbase: A database curates experimentally validated microrna-target interactions. Nucleic Acids Res. 2011, 39, D163–D169. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Van der Net, J.B.; Janssens, A.C.; Eijkemans, M.J.; Kastelein, J.J.; Sijbrands, E.J.; Steyerberg, E.W. Cox proportional hazards models have more statistical power than logistic regression models in cross-sectional genetic association studies. Eur. J. Hum. Genet. 2008, 16, 1111–1116. [Google Scholar] [CrossRef]
- Song, Y.X.; Sun, J.X.; Zhao, J.H.; Yang, Y.C.; Shi, J.X.; Wu, Z.H.; Chen, X.W.; Gao, P.; Miao, Z.F.; Wang, Z.N. Non-coding rnas participate in the regulatory network of cldn4 via cerna mediated mirna evasion. Nat. Commun. 2017, 8, 289. [Google Scholar] [CrossRef]
- Meng, Q.; Li, S.; Liu, Y.; Zhang, S.; Jin, J.; Zhang, Y.; Guo, C.; Liu, B.; Sun, Y. Circular RNA circscaf11 accelerates the glioma tumorigenesis through the mir-421/sp1/vegfa axis. Mol. Ther. Nucleic Acids 2019, 17, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, X.; Wang, W.; Xie, H.; Li, J.; Hu, Z.; Zheng, Z.; Li, H.; Teng, L. Lncrna cdkn2bas predicts poor prognosis in patients with hepatocellular carcinoma and promotes metastasis via the mir-153-5p/arhgap18 signaling axis. Aging 2018, 10, 3371–3381. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, B.; Ran, P.; Wang, L. Identification of cerna network based on a RNA-seq shows prognostic lncrna biomarkers in human lung adenocarcinoma. Oncol. Lett. 2018, 16, 5697–5708. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhong, Z.; Lv, M.; Shu, J.; Tian, Q.; Chen, J. Comprehensive analysis of differentially expressed profiles of lncrnas and circrnas with associated co-expression and cerna networks in bladder carcinoma. Oncotarget 2016, 7, 47186–47200. [Google Scholar] [CrossRef]
- Zheng, M.J.; Gou, R.; Zhang, W.C.; Nie, X.; Wang, J.; Gao, L.L.; Liu, J.J.; Li, X.; Lin, B. Screening of prognostic biomarkers for endometrial carcinoma based on a cerna network. PeerJ 2018, 6, e6091. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, S.; Ji, D.; Qian, W.; Wang, Q.; Li, J.; Gu, J.; Peng, W.; Hu, T.; Ji, B.; et al. Construction of a cerna network reveals potential lncrna biomarkers in rectal adenocarcinoma. Oncol. Rep. 2018, 39, 2101–2113. [Google Scholar] [CrossRef]
- McConnell, B.B.; Vertino, P.M. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein tms1. Cancer Res. 2000, 60, 6243–6247. [Google Scholar]
- Conway, K.E.; McConnell, B.B.; Bowring, C.E.; Donald, C.D.; Warren, S.T.; Vertino, P.M. Tms1, a novel proapoptotic caspase recruitment domain protein, is a target of methylation-induced gene silencing in human breast cancers. Cancer Res. 2000, 60, 6236–6242. [Google Scholar]
- Mhyre, A.J.; Marcondes, A.M.; Spaulding, E.Y.; Deeg, H.J. Stroma-dependent apoptosis in clonal hematopoietic precursors correlates with expression of pycard. Blood 2009, 113, 649–658. [Google Scholar] [CrossRef]
- Momeni, M.; Reza Mirzaei, M.; Zainodini, N.; Hassanshahi, G.; Arababadi, M.K. Mir-143 induces expression of aim2 and asc in jurkat cell line. Iran. J. Immunol. 2013, 10, 103–109. [Google Scholar]
- Li, Y.; Shao, H.; Da, Z.; Pan, J.; Fu, B. High expression of slc38a1 predicts poor prognosis in patients with de novo acute myeloid leukemia. J. Cell. Physiol. 2019, 234, 20322–20328. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, P.; Gao, H.F.; Qian, J.X.; Yuan, L.Y.; Wang, J.J. Overexpression of slc38a1 is associated with poorer prognosis in chinese patients with gastric cancer. BMC Gastroenterol. 2014, 14, 70. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.F.; Xie, W.; Chen, S.Q.; Wang, X.K.; Liu, Q.; Pan, X.K.; Su, F.; Feng, M.H. Slc38a1 promotes proliferation and migration of human colorectal cancer cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2017, 37, 30–36. [Google Scholar] [CrossRef] [PubMed]
- De Groot, J.S.; Moelans, C.B.; Elias, S.G.; Jo Fackler, M.; van Domselaar, R.; Suijkerbuijk, K.P.; Witkamp, A.J.; Sukumar, S.; van Diest, P.J.; van der Wall, E. DNA promoter hypermethylation in nipple fluid: A potential tool for early breast cancer detection. Oncotarget 2016, 7, 24778–24791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- De Groot, J.S.; Pan, X.; Meeldijk, J.; van der Wall, E.; van Diest, P.J.; Moelans, C.B. Validation of DNA promoter hypermethylation biomarkers in breast cancer—A short report. Cell. Oncol. 2014, 37, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Li, J.; Xin, J.; Wu, J.; Guo, J.; Zhang, L.; Jiang, L.; Zhang, W.; Yang, Z.; Li, L. Methylation profile of single hepatocytes derived from hepatitis b virus-related hepatocellular carcinoma. PLoS ONE 2011, 6, e19862. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liu, X.; Wang, Z.; She, X.; Zeng, X.; Deng, M.; Liao, Q.; Guo, X.; Wang, R.; Li, X.; et al. Interaction of hsa-mir-381 and glioma suppressor lrrc4 is involved in glioma growth. Brain Res. 2011, 1390, 21–32. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Z.; Liu, Q.; Liu, X.; Wu, M.; Li, G. Disturbing mir-182 and -381 inhibits brd7 transcription and glioma growth by directly targeting lrrc4. PLoS ONE 2014, 9, e84146. [Google Scholar] [CrossRef]
- Zekri, A.R.; Hassan, Z.K.; Bahnassy, A.A.; Eldahshan, D.H.; El-Rouby, M.N.; Kamel, M.M.; Hafez, M.M. Gene expression profiling of non-hodgkin lymphomas. Asian Pac. J. Cancer Prev. 2013, 14, 4393–4398. [Google Scholar] [CrossRef]
- Xiao, L.; Tu, C.; Chen, S.; Yu, Z.; Lei, Q.; Wang, Z.; Xu, G.; Wu, M.; Li, G. Lrrc4 haplotypes are associated with pituitary adenoma in a chinese population. Med. Oncol. 2014, 31, 888. [Google Scholar] [CrossRef]
- Zhou, X.J.; Wang, Y.; Zhang, L.J.; Chen, J.H. Expression and clinical significance of lrrc4 in benign and malignant nasopharyngeal diseases. Genet. Mol. Res. 2015, 14, 16403–16409. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, C.; Bacher, U.; Kohlmann, A.; Schindela, S.; Alpermann, T.; Kern, W.; Schnittger, S.; Haferlach, T. Cdkn1b, encoding the cyclin-dependent kinase inhibitor 1b (p27), is located in the minimally deleted region of 12p abnormalities in myeloid malignancies and its low expression is a favorable prognostic marker in acute myeloid leukemia. Haematologica 2011, 96, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Han, Y.; Feng, Y.; Xia, L.; Hsu, C.; Sheng, X.; Pooler, L.C.; Patel, Y.; Kolonel, L.N.; Carter, E.; et al. Genome-wide testing of putative functional exonic variants in relationship with breast and prostate cancer risk in a multiethnic population. PLoS Genet. 2013, 9, e1003419. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.; Chen, Q.; Wang, Q.; Yang, L.; Lv, D.; Jin, G.; Liu, J.; Li, B.; Fei, X. Atf-3/mir-590/golph3 signaling pathway regulates proliferation of breast cancer. BMC Cancer 2018, 18, 255. [Google Scholar] [CrossRef]
- Li, X.; Xu, M.; Ding, L.; Tang, J. Mir-27a: A novel biomarker and potential therapeutic target in tumors. J. Cancer 2019, 10, 2836–2848. [Google Scholar] [CrossRef]
- Gu, B.; Wang, J.; Song, Y.; Wang, Q.; Wu, Q. Microrna-383 regulates cell viability and apoptosis by mediating wnt/β-catenin signaling pathway in non-small cell lung cancer. J. Cell. Biochem. 2018, 120, 7918–7926. [Google Scholar] [CrossRef]
- Krajewska, J.B.; Fichna, J.; Mosinska, P. One step ahead: Mirna-34 in colon cancer-future diagnostic and therapeutic tool? Crit. Rev. Oncol. Hematol. 2018, 132, 1–8. [Google Scholar] [CrossRef]
- Feng, L.; Houck, J.R.; Lohavanichbutr, P.; Chen, C. Transcriptome analysis reveals differentially expressed lncrnas between oral squamous cell carcinoma and healthy oral mucosa. Oncotarget 2017, 8, 31521–31531. [Google Scholar] [CrossRef]
- Zhou, F.; Wang, D.; Wei, W.; Chen, H.; Shi, H.; Zhou, N.; Wu, L.; Peng, R. Comprehensive profiling of circular RNA expressions reveals potential diagnostic and prognostic biomarkers in multiple myeloma. BMC Cancer 2020, 20, 40. [Google Scholar] [CrossRef]
- Wangxia, L.V.; Fang, Y.; Liu, Y.; Zhao, Y.; Shi, Z.; Zhong, H. Circular RNA arhgap26 is over-expressed and its downregulation inhibits cell proliferation and promotes cell apoptosis in gastric cancer cells. Saudi J. Gastroenterol. Off. J. Saudi Gastroenterol. Assoc. 2019, 25, 119–125. [Google Scholar]
- Yang, Q.; Tang, Y.; Tang, C.; Cong, H.; Wang, X.; Shen, X.; Ju, S. Diminished linc00173 expression induced mir-182-5p accumulation promotes cell proliferation, migration and apoptosis inhibition via ager/nf-kappab pathway in non-small-cell lung cancer. Am. J. Transl. Res. 2019, 11, 4248–4262. [Google Scholar] [PubMed]
- Han, P.; Li, J.W.; Zhang, B.M.; Lv, J.C.; Li, Y.M.; Gu, X.Y.; Yu, Z.W.; Jia, Y.H.; Bai, X.F.; Li, L.; et al. The lncrna crnde promotes colorectal cancer cell proliferation and chemoresistance via mir-181a-5p-mediated regulation of wnt/β-catenin signaling. Mol. Cancer 2017, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Tao, W.; Ni, S.; Chen, Q. Upregulation of lncrna snorna host gene 6 regulates nuak family snf1-like kinase-1 expression by competitively binding microrna-125b and interacting with snail1/2 in bladder cancer. J. Cell. Biochem. 2019, 120, 357–367. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alive (n = 54) | Dead (n = 97) | Total (n = 151) | |
|---|---|---|---|
| Gender | |||
| Female | 24(44.4%) | 44(45.4%) | 68(45.0%) |
| Male | 30(55.6%) | 53(54.6%) | 83 (55.0%) |
| FAB Classification | |||
| M0 | 5(9.3%) | 10(10.3%) | 15(9.9%) |
| M1 | 11(20.4%) | 24(24.7%) | 35(23.2%) |
| M2 | 14(25.9%) | 24(24.7%) | 38(25.2%) |
| M3 | 11(20.4%) | 4(4.1%) | 15(9.9%) |
| M4 | 8(14.8%) | 21(21.6%) | 29(19.2%) |
| M5 | 5(9.3%) | 10(10.3%) | 15(9.9%) |
| M6 | 2(1.3%) | 2(1.3%) | |
| M7 | 1(0.7%) | 1(0.7%) | |
| Not Classified | 1(0.7%) | 1(0.7%) | |
| Cytogenetic Risk Group | |||
| Favorable | 19(35.2%) | 12(12.4%) | 31(20.6%) |
| Intermediate | 29(53.7%) | 53(54.6%) | 82(54.3%) |
| Poor | 6(11.1%) | 30(30.9%) | 36(23.8%) |
| Missing data | 2(2.1%) | 2(1.3%) |
| Ontology | Term | Description | Count | Adj. p-Value |
|---|---|---|---|---|
| BP | GO:0043312 | neutrophil degranulation | 56 | 5.38148E-26 |
| BP | GO:0001819 | positive regulation of cytokine production | 30 | 9.621E-07 |
| BP | GO:0032640 | tumor necrosis factor production | 15 | 0.000148136 |
| BP | GO:0032757 | positive regulation of interleukin-8 production | 8 | 0.000698632 |
| BP | GO:0032609 | interferon-γ production | 11 | 0.001628489 |
| CC | GO:0034774 | secretory granule lumen | 33 | 1.18956E-13 |
| CC | GO:0060205 | cytoplasmic vesicle lumen | 33 | 1.98605E-13 |
| CC | GO:0101002 | ficolin-1-rich granule | 23 | 1.59718E-11 |
| CC | GO:0005766 | primary lysosome | 14 | 2.12213E-05 |
| CC | GO:0005802 | trans-Golgi network | 16 | 0.000131017 |
| MF | GO:0030246 | carbohydrate binding | 17 | 0.007065521 |
| MF | GO:0008329 | signaling pattern recognition receptor activity | 5 | 0.007065521 |
| MF | GO:0038187 | pattern recognition receptor activity | 5 | 0.007065521 |
| TermID | Description | Count | Adj. p-Value |
|---|---|---|---|
| hsa04640 | Hematopoietic cell lineage | 11 | 0.003548969 |
| hsa04612 | Antigen processing and presentation | 9 | 0.008063516 |
| hsa05150 | Staphylococcus aureus infection | 9 | 0.026866028 |
| hsa04142 | Lysosome | 10 | 0.030064746 |
| hsa04145 | Phagosome | 11 | 0.034084109 |
| hsa05152 | Tuberculosis | 12 | 0.034084109 |
| hsa05132 | Salmonella infection | 13 | 0.042405443 |
| Gene | Univariate Cox | Multivariate Cox | |
|---|---|---|---|
| HR | p-Value | p-Value | |
| ZMAT1 | 0.334297693 | 0.00048 | 0.0412 |
| MANSC1 | 2.233815416 | 0.0021 | 0.0775 |
| TM6SF1 | 0.490252653 | 0.00312 | 0.0443 |
| PYCARD | 3.194180152 | 0.00478 | 0.0081 |
| RAB31 | 1.426030724 | 0.00971 | |
| SLC38A1 | 1.630159352 | 0.01083 | 0.0036 |
| IGF2R | 1.466708157 | 0.01339 | |
| CLEC7A | 1.315460822 | 0.01363 | |
| UBXN11 | 1.805081888 | 0.01387 | |
| SLC8A1 | 1.587431529 | 0.01947 | |
| NRIP1 | 1.521500351 | 0.01983 | |
| LRRC4 | 0.449053605 | 0.02275 | 0.0812 |
| SLC9A7 | 1.594442485 | 0.02472 | |
| FGD4 | 1.34192418 | 0.04989 | |
| Type | Hub RNA | |||||
|---|---|---|---|---|---|---|
| mRNA | TM6SF1 | ZMAT1 | MANSC1 | PYCARD | SLC38A1 | LRRC4 |
| miRNA | hsa-mir-590-3p | hsa-mir-27a-5p | hsa-mir-383-3p | hsa-mir-34c-3p | ||
| hsa-mir-215-3p | hsa-mir-506-5p | hsa-mir-182-5p | hsa-mir-17-3p | |||
| circRNA | hsa_circ_0003602 | hsa_circ_0005571 | hsa_circ_0074371 | |||
| hsa_circ_0007609 | hsa_circ_0012152 | hsa_circ_0001247 | ||||
| lncRNA | PCBP1-AS1 | ASH1L-IT1 | ITCH-IT1 | CYP1B1-AS1 | C5orf58 | GAS5 |
| SNHG1 | TPTEP1 | FGD5-AS1 | MIAT | GUSBP11 | LINC00342 | |
| FOXP1-AS1 | CRNDE | SNHG6 | MIR210HG | SCARNA2 | MYB-AS1 | |
| MALAT1 | AC138035.1 | LINC00173 | TUG1 | OIP5-AS1 | FAM157B | |
| HOTAIRM1 | ANKRD44-IT1 | SNHG3 | HCG25 | HMGA1P4 | GAS6-AS1 | |
| H19 | ATP1B3-AS1 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Su, Y.; Wang, S.; Liu, Y.; Jin, L.; Wan, Q.; Liu, Y.; Li, C.; Sang, X.; Yang, L.; et al. Identification of circRNA-lncRNA-miRNA-mRNA Competitive Endogenous RNA Network as Novel Prognostic Markers for Acute Myeloid Leukemia. Genes 2020, 11, 868. https://doi.org/10.3390/genes11080868
Cheng Y, Su Y, Wang S, Liu Y, Jin L, Wan Q, Liu Y, Li C, Sang X, Yang L, et al. Identification of circRNA-lncRNA-miRNA-mRNA Competitive Endogenous RNA Network as Novel Prognostic Markers for Acute Myeloid Leukemia. Genes. 2020; 11(8):868. https://doi.org/10.3390/genes11080868
Chicago/Turabian StyleCheng, Yaqi, Yaru Su, Shoubi Wang, Yurun Liu, Lin Jin, Qi Wan, Ying Liu, Chaoyang Li, Xuan Sang, Liu Yang, and et al. 2020. "Identification of circRNA-lncRNA-miRNA-mRNA Competitive Endogenous RNA Network as Novel Prognostic Markers for Acute Myeloid Leukemia" Genes 11, no. 8: 868. https://doi.org/10.3390/genes11080868
APA StyleCheng, Y., Su, Y., Wang, S., Liu, Y., Jin, L., Wan, Q., Liu, Y., Li, C., Sang, X., Yang, L., Liu, C., & Wang, Z. (2020). Identification of circRNA-lncRNA-miRNA-mRNA Competitive Endogenous RNA Network as Novel Prognostic Markers for Acute Myeloid Leukemia. Genes, 11(8), 868. https://doi.org/10.3390/genes11080868