Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations

 ,
,  , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Introduction
2. Pathogenesis and Histology—Osteoarthritis as a Whole Joint Disease
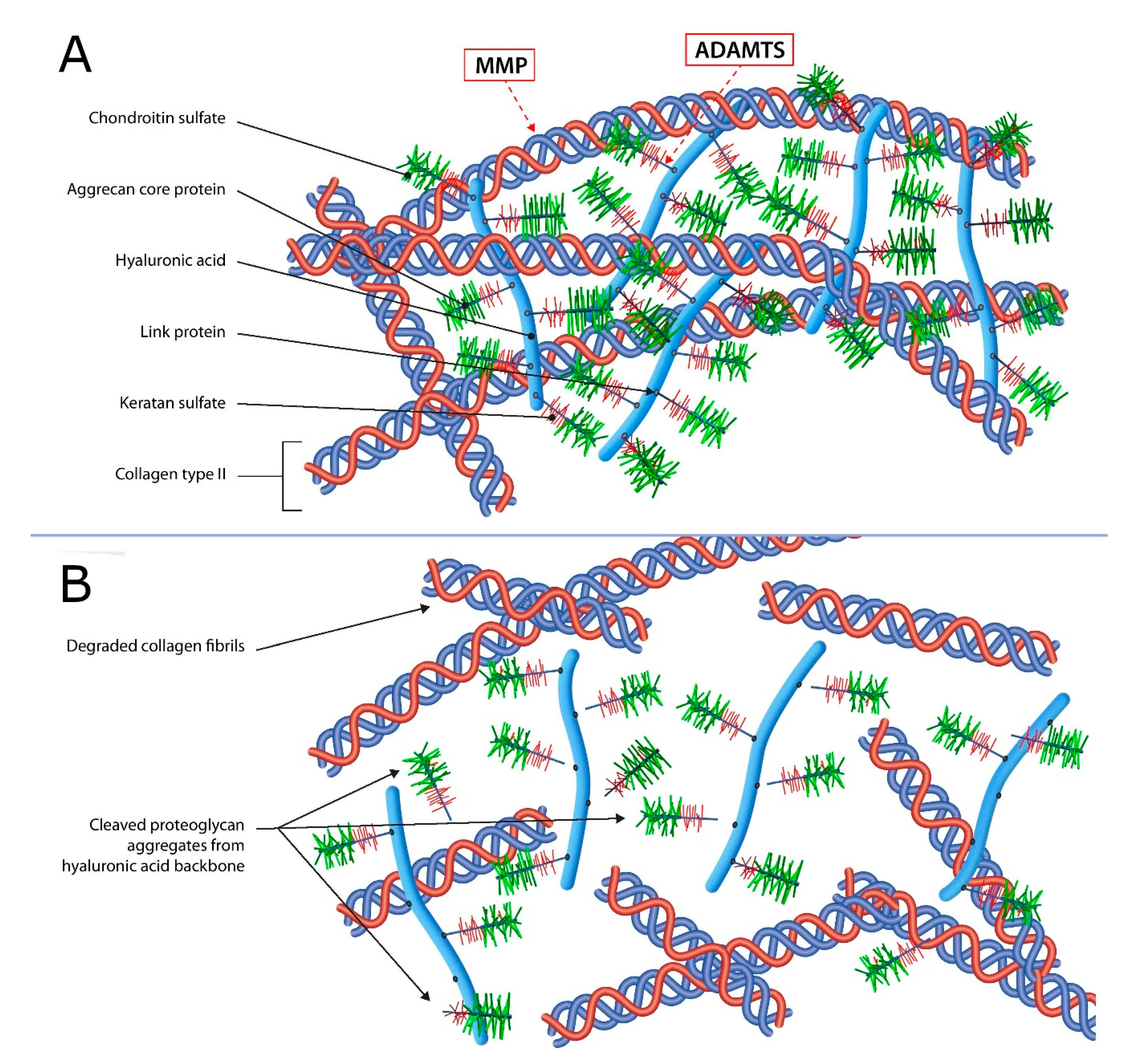
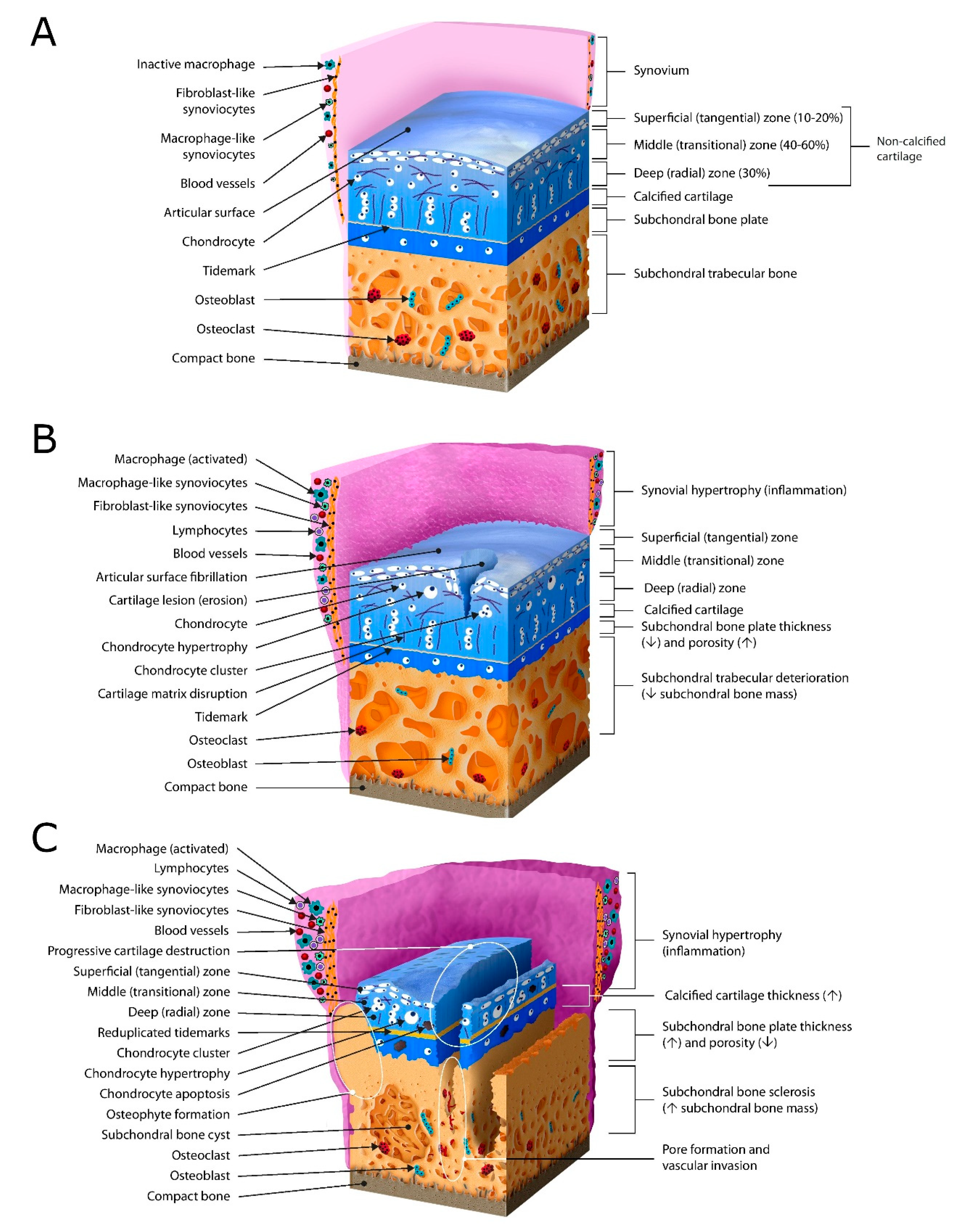
2.1. Articular Cartilage
2.2. Subchondral Bone
2.3. Synovium
2.4. Infrapatellar Fat Pad, Menisci, Periarticular Muscles, Ligaments and Tendons
2.5. The Role of Cytokines, Chemokines, Mirna, Gene Expression and Other Molecules
2.5.1. Cytokines Involved in OA Signalization
2.5.2. Adipokines
2.5.3. Genetics and Epigenetics of OA
2.5.4. The Role of miRNAs in Epigenetic Regulation of OA
2.5.5. The Mitochondrial Genetics and Epigenetics in Osteoarthritis
3. State-Of-The-Art Non-Operative Therapeutic Consideration
3.1. Platelet-Rich Plasma
3.2. Bone Marrow Mesenchymal Stem Cells
- Interference with the underlying pathological or inflammatory process via immune modulation [193]. This effect is expressed through the suppression of Th1 and up-regulation of Th2 cells, inhibiting IFN-y production by interaction with NK cells, switching the phenotype macrophages from proinflammatory M1 to M2, and reducing the antibody production of B-lymphocytes [192];
- MiRNA secretion, regulating the gene expression of the surrounding cells [192].
3.3. Intra-articular Application of Autologous Microfragmented Adipose Tissue with Stromal Vascular Fraction
3.4. Extracellular Vesicles
4. Future Strategies for OA Management
4.1. Phenotyping OA
4.2. Human-Recombinant Fibroblast Growth Factor 18 (Sprifermin)
4.3. Bone Morphogenetic Protein 7 (BMP-7)
4.4. Monoclonal Antibodies
4.5. Gene Therapy of OA
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bortoluzzi, A.; Furini, F.; Scirè, C.A. Osteoarthritis and its management—Epidemiology, nutritional aspects and environmental factors. Autoimmun. Rev. 2018, 17, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Mabey, T.; Honsawek, S. Cytokines as biochemical markers for knee osteoarthritis. World J. Orthop. 2015, 6, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.E. Osteoarthritis year in review 2017: Clinical. Osteoarthr. Cartil. 2018, 26, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Loeser, R.F.; Goldring, S.R.; Scanzello, C.R.; Goldring, M.B. Osteoarthritis: A disease of the joint as an organ. Arthritis Rheum. 2012, 64, 1697–1707. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Barr, A.J.; Cicuttini, F.M.; Conaghan, P.G.; Cooper, C.; Goldring, M.B.; Goldring, S.R.; Jones, G.; Teichtahl, A.J.; Pelletier, J.P. Osteoarthritis. Nat. Rev. Dis. Prim. 2016, 2, 1–18. [Google Scholar] [CrossRef]
- Swingler, T.E.; Niu, L.; Smith, P.; Paddy, P.; Le, L.; Barter, M.J.; Young, D.A.; Clark, I.M. The function of microRNAs in cartilage and osteoarthritis. Clin. Exp. Rheumatol. 2019, 37, 40–47. [Google Scholar]
- Ilas, D.C.; Churchman, S.M.; McGonagle, D.; Jones, E. Targeting subchondral bone mesenchymal stem cell activities for intrinsic joint repair in osteoarthritis. Futur. Sci. OA 2017, 3, FSO228. [Google Scholar] [CrossRef]
- Hunter, D.J.; Bierma-Zeinstra, S. Osteoarthritis. Lancet 2019, 393, 1745–1759. [Google Scholar] [CrossRef]
- Carlson, A.K.; Rawle, R.A.; Wallace, C.W.; Brooks, E.G.; Adams, E.; Greenwood, M.C.; Olmer, M.; Lotz, M.K.; Bothner, B.; June, R.K. Characterization of synovial fluid metabolomic phenotypes of cartilage morphological changes associated with osteoarthritis. Osteoarthr. Cartil. 2019, 27, 1174–1184. [Google Scholar] [CrossRef]
- Nguyen, U.-S.D.T.; Zhang, Y.; Zhu, Y.; Niu, J.; Zhang, B.; Felson, D.T. Increasing Prevalence of Knee Pain and Symptomatic Knee Osteoarthritis: Survey and Cohort Data. Ann. Intern. Med. 2011, 155, 725–732. [Google Scholar] [CrossRef]
- Zhang, Y.; Jordan, J.M. Epidemiology of Osteoarthritis. Clin. Geriatr. Med. 2010, 26, 355–369. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1545–1602. [CrossRef]
- Prince, M.J.; Wu, F.; Guo, Y.; Gutierrez Robledo, L.M.; O’Donnell, M.; Sullivan, R.; Yusuf, S. The burden of disease in older people and implications for health policy and practice. Lancet 2015, 385, 549–562. [Google Scholar] [CrossRef]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthritis an age-related disease? Best Pract. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T. Osteoarthritis as a disease of mechanics. Osteoarthr. Cartil. 2013, 21, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Vina, E.R.; Kwoh, C.K. Epidemiology of osteoarthritis. Curr. Opin. Rheumatol. 2018, 30, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.C.; Coggon, D. HIP osteoarthritis and work. Best Pract. Res. Clin. Rheumatol. 2015, 29, 462–482. [Google Scholar] [CrossRef]
- Ezzat, A.M.; Li, L.C. Occupational Physical Loading Tasks and Knee Osteoarthritis: A Review of the Evidence. Physiother. Can. 2014, 66, 91–107. [Google Scholar] [CrossRef]
- Driban, J.B.; Hootman, J.M.; Sitler, M.R.; Harris, K.P.; Cattano, N.M. Is Participation in Certain Sports Associated With Knee Osteoarthritis? A Systematic Review. J. Athl. Train. 2017, 52, 497–506. [Google Scholar] [CrossRef]
- Berenbaum, F.; Wallace, I.J.; Lieberman, D.E.; Felson, D.T. Modern-day environmental factors in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2018, 14, 674–681. [Google Scholar] [CrossRef]
- Hame, S.L.; Alexander, R.A. Knee osteoarthritis in women. Curr. Rev. Musculoskelet. Med. 2013, 6, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Long, H.; Zeng, X.; Liu, Q.; Wang, H.; Vos, T.; Hou, Y.; Lin, C.; Qiu, Y.; Wang, K.; Xing, D.; et al. Burden of osteoarthritis in China, 1990–2017: Findings from the Global Burden of Disease Study 2017. Lancet Rheumatol. 2020, 2, e164–e172. [Google Scholar] [CrossRef]
- Wang, H.; Bai, J.; He, B.; Hu, X.; Liu, D. Osteoarthritis and the risk of cardiovascular disease: A meta-analysis of observational studies. Sci. Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wallace, I.J.; Worthington, S.; Felson, D.T.; Jurmain, R.D.; Wren, K.T.; Maijanen, H.; Woods, R.J.; Lieberman, D.E. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl. Acad. Sci. USA 2017, 114, 9332–9336. [Google Scholar] [CrossRef]
- Veronese, N.; Cereda, E.; Maggi, S.; Luchini, C.; Solmi, M.; Smith, T.; Denkinger, M.; Hurley, M.; Thompson, T.; Manzato, E.; et al. Osteoarthritis and mortality: A prospective cohort study and systematic review with meta-analysis. Semin. Arthritis Rheum. 2016, 46, 160–167. [Google Scholar] [CrossRef]
- Bianco, D.; Todorov, A.; Čengić, T.; Pagenstert, G.; Schären, S.; Netzer, C.; Hügle, T.; Geurts, J. Alterations of Subchondral Bone Progenitor Cells in Human Knee and Hip Osteoarthritis Lead to a Bone Sclerosis Phenotype. Int. J. Mol. Sci. 2018, 19, 475. [Google Scholar] [CrossRef]
- Reynard, L.N.; Barter, M.J. Osteoarthritis year in review 2019: Genetics, genomics and epigenetics. Osteoarthr. Cartil. 2020, 28, 275–284. [Google Scholar] [CrossRef]
- Rice, S.J.; Beier, F.; Young, D.A.; Loughlin, J. Interplay between genetics and epigenetics in osteoarthritis. Nat. Rev. Rheumatol. 2020, 16, 268–281. [Google Scholar] [CrossRef]
- Emery, C.A.; Whittaker, J.L.; Mahmoudian, A.; Lohmander, L.S.; Roos, E.M.; Bennell, K.L.; Toomey, C.M.; Reimer, R.A.; Thompson, D.; Ronsky, J.L.; et al. Establishing outcome measures in early knee osteoarthritis. Nat. Rev. Rheumatol. 2019, 15, 438–448. [Google Scholar] [CrossRef]
- Xia, B.; Chen, D.; Zhang, J.; Hu, S.; Jin, H.; Tong, P. Osteoarthritis Pathogenesis: A Review of Molecular Mechanisms. Calcif. Tissue Int. 2014, 95, 495–505. [Google Scholar] [CrossRef]
- Goldring, S.R.; Goldring, M.B. Changes in the osteochondral unit during osteoarthritis: Structure, function and cartilage bone crosstalk. Nat. Rev. Rheumatol. 2016, 12, 632–644. [Google Scholar] [CrossRef]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic Science of Articular Cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Primorac, D.; Stover, M.L.; Clark, S.H.; Rowe, D.W. Molecular basis of nanomelia, a heritable chondrodystrophy of chicken. Matrix Biol. 1994, 14, 297–305. [Google Scholar] [CrossRef]
- Primorac, D. Reduced Type II collagen mRNA in nanomelic cultured chondrocytes: An example of extracellular matrix/collagen feedback regulation? Croat. Med. J. 1995, 36, 85–92. [Google Scholar]
- Primorac, D.; Johnson, C.; Lawrence, J.; McKinstry, M.; Stover, M.; Schanfield, M.; Andelinović, S.; Tadić, T.; Rowe, D. Premature termination codon in the aggrecan gene of nanomelia and its influence on mRNA transport and stability. Croat. Med. J. 1999, 40, 528–532. [Google Scholar]
- Houard, X.; Goldring, M.B.; Berenbaum, F. Homeostatic Mechanisms in Articular Cartilage and Role of Inflammation in Osteoarthritis. Curr. Rheumatol. Rep. 2013, 15, 375. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G. The Effect of Mechanical Loading on Articular Cartilage. J. Funct. Morphol. Kinesiol. 2016, 1, 154–161. [Google Scholar] [CrossRef]
- Jacobs, C.R.; Huang, H.; Kwon, R.Y. Introduction to Cell Mechanics and Mechanobiology; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Vanwanseele, B.; Eckstein, F.; Knecht, H.; Spaepen, A.; Stüssis, E. Longitudinal Analysis of Cartilage Atrophy in the Knees of Patients with Spinal Cord Injury. Arthritis Rheum. 2003, 48, 3377–3381. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, J.C.; Mandalia, V.; Toms, A.; Winlove, C.P.; Brasselet, S. Collagen reorganization in cartilage under strain probed by polarization sensitive second harmonic generation microscopy. J. R. Soc. Interface 2019, 16, 20180611. [Google Scholar] [CrossRef]
- Mansfield, J.C.; Bell, J.S.; Winlove, C.P. The micromechanics of the superficial zone of articular cartilage. Osteoarthr. Cartil. 2015, 23, 1806–1816. [Google Scholar] [CrossRef] [PubMed]
- Korhonen, R.K.; Julkunen, P.; Wilson, W.; Herzog, W. Importance of Collagen Orientation and Depth-Dependent Fixed Charge Densities of Cartilage on Mechanical Behavior of Chondrocytes. J. Biomech. Eng. 2008, 130, 021003. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.; Driessen, N.J.B.; van Donkelaar, C.C.; Ito, K. Prediction of collagen orientation in articular cartilage by a collagen remodeling algorithm. Osteoarthr. Cartil. 2006, 14, 1196–1202. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.P.; Kirk, T.B.; Zheng, M.H. Study of the collagen structure in the superficial zone and physiological state of articular cartilage using a 3D confocal imaging technique. J. Orthop. Surg. Res. 2008, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, J.C.; Peter Winlove, C. A multi-modal multiphoton investigation of microstructure in the deep zone and calcified cartilage. J. Anat. 2012, 220, 405–416. [Google Scholar] [CrossRef]
- Ruhlen, R.; Marberry, K. The chondrocyte primary cilium. Osteoarthr. Cartil. 2014, 22, 1071–1076. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann. N. Y Acad. Sci. 2010, 1192, 230–237. [Google Scholar] [CrossRef]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef]
- Pratta, M.A.; Su, J.L.; Leesnitzer, M.A.; Struglics, A.; Larsson, S.; Lohmander, L.S.; Kumar, S. Development and characterization of a highly specific and sensitive sandwich ELISA for detection of aggrecanase-generated aggrecan fragments. Osteoarthr. Cartil. 2006, 14, 702–713. [Google Scholar] [CrossRef]
- Loeser, R.F. Molecular mechanisms of cartilage destruction: Mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006, 54, 1357–1360. [Google Scholar] [CrossRef]
- Parrish, A.R. Matrix Metalloproteinases and Tissue Remodeling in Health and Disease: Target Tissues and Therapy, 1st ed.; Elsevier: San Diego, CA, USA, 2017; p. 310. [Google Scholar]
- Rolauffs, B.; Williams, J.M.; Aurich, M.; Grodzinsky, A.J.; Kuettner, K.E.; Cole, A.A. Proliferative Remodeling of the Spatial Organization of Human Superficial Chondrocytes Distant From Focal Early Osteoarthritis. Arthritis Rheum. Off. J. Am. Coll. Rheum. 2010, 62, 489–498. [Google Scholar] [CrossRef]
- Glyn-Jones, S.; Palmer, A.J.R.; Agricola, R.; Price, A.J.; Vincent, T.L.; Weinans, H.; Carr, A.J. Osteoarthritis. Lancet 2015, 386, 376–387. [Google Scholar] [CrossRef]
- Zhen, G.; Cao, X. Targeting TGFβ signaling in subchondral bone and articular cartilage homeostasis. Trends Pharmacol. Sci. 2014, 35, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yin, J.; Gao, J.; Cheng, T.S.; Pavlos, N.J.; Zhang, C.; Zheng, M.H. Subchondral bone in osteoarthritis: Insight into risk factors and microstructural changes. Arthritis Res. Ther. 2013, 15, 223. [Google Scholar] [CrossRef]
- Funck-Brentano, T.; Cohen-Solal, M. Subchondral bone and osteoarthritis. Curr. Opin. Rheumatol. 2015, 27, 420–426. [Google Scholar] [CrossRef]
- Sanchez, C.; Pesesse, L.; Gabay, O.; Delcour, J.P.; Msika, P.; Baudouin, C.; Henrotin, Y.E. Regulation of subchondral bone osteoblast metabolism by cyclic compression. Arthritis Rheum. 2012, 64, 1193–1203. [Google Scholar] [CrossRef]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Goldring, S.R. Role of Bone in Osteoarthritis Pathogenesis. Med. Clin. N. Am. 2009, 93, 25–35. [Google Scholar] [CrossRef]
- Driban, J.B.; Tassinari, A.; Lo, G.H.; Price, L.L.; Schneider, E.; Lynch, J.A.; Eaton, C.B.; McAlindon, T.E. Bone marrow lesions are associated with altered trabecular morphometry. Osteoarthr. Cartil. 2012, 20, 1519–1526. [Google Scholar] [CrossRef]
- Bowes, M.A.; McLure, S.W.; Wolstenholme, C.B.; Vincent, G.R.; Williams, S.; Grainger, A.; Conaghan, P.G. Osteoarthritic bone marrow lesions almost exclusively colocate with denuded cartilage: A 3D study using data from the Osteoarthritis Initiative. Ann. Rheum. Dis. 2016, 75, 1852–1857. [Google Scholar] [CrossRef] [PubMed]
- Cotofana, S.; Wyman, B.T.T.; Benichou, O.; Dreher, D.; Nevitt, M.; Gardiner, J.; Wirth, W.; Hitzl, W.; Kwoh, C.K.K.; Eckstein, F.; et al. Relationship between knee pain and the presence, location, size and phenotype offemorotibial denuded areas of subchondral bone as visualized by MRI. Osteoarthr. Cartil. 2013, 21, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Crema, M.D.; Roemer, F.W.; Zhu, Y.; Marra, M.D.; Niu, J.; Zhang, Y.; Lynch, J.A.; Javaid, M.K.; Lewis, C.E.; El-Khoury, G.Y.; et al. Subchondral cystlike lesions develop longitudinally in areas of bone marrow edema-like lesions in patients with or at risk for knee osteoarthritis: Detection with MR imaging—The MOST study. Radiology 2010, 256, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Li, P.; Zhu, S.; Bi, R. Comparison of early-stage changes of osteoarthritis in cartilage and subchondral bone between two different rat models. PeerJ 2020, 8, e8934. [Google Scholar] [CrossRef] [PubMed]
- Crema, M.D.D.; Cibere, J.; Sayre, E.C.C.; Roemer, F.W.W.; Wong, H.; Thorne, A.; Singer, J.; Esdaile, J.M.M.; Marra, M.D.D.; Kopec, J.A.A.; et al. The relationship between subchondral sclerosis detected with MRI and cartilage loss in a cohort of subjects with knee pain: The knee osteoarthritis progression (KOAP) study. Osteoarthr. Cartil. 2014, 22, 540–546. [Google Scholar] [CrossRef]
- Wenham, C.Y.J.; Conaghan, P.G. The role of synovitis in osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2010, 2, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.A.; Gastelum, N.S.; Nguyen, Q.T.; Schumacher, B.L.; Sah, R.L. Boundary lubrication of articular cartilage: Role of synovial fluid constituents. Arthritis Rheum. 2007, 56, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Griffin, T.M.; Scanzello, C.R. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin. Exp. Rheumatol. 2019, 37, 57–63. [Google Scholar]
- Belluzzi, E.; Stocco, E.; Pozzuoli, A.; Granzotto, M.; Porzionato, A.; Vettor, R.; De Caro, R.; Ruggieri, P.; Ramonda, R.; Rossato, M.; et al. Contribution of Infrapatellar Fat Pad and Synovial Membrane to Knee Osteoarthritis Pain. Biomed. Res. Int. 2019, 2019, 1–18. [Google Scholar] [CrossRef]
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010, 6, 625–635. [Google Scholar] [CrossRef]
- Xie, J.; Huang, Z.; Yu, X.; Zhou, L.; Pei, F. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. Cytokine Growth Factor Rev. 2019, 46, 36–44. [Google Scholar] [CrossRef]
- HüGle, T.; Geurts, J. What drives osteoarthritis?-synovial versus subchondral bone pathology. Rheumatology 2016, 56, 1461–1471. [Google Scholar] [CrossRef]
- Ene, R.; Diana Sinescu, R.; Ene, P.; Mihaela Cîrstoiu, M.; Cătălin Cîrstoiu, F. Synovial inflammation in patients with different stages of knee osteoarthritis. Rom. J. Morphol. Embryol. 2015, 56, 169–173. [Google Scholar] [PubMed]
- de Lange-Brokaar, B.J.E.; Ioan-Facsinay, A.; van Osch, G.J.V.M.; Zuurmond, A.-M.; Schoones, J.; Toes, R.E.M.; Huizinga, T.W.J.; Kloppenburg, M. Synovial inflammation, immune cells and their cytokines in osteoarthritis: A review. Osteoarthr. Cartil. 2012, 20, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Ayral, X.; Pickering, E.H.; Woodworth, T.G.; Mackillop, N.; Dougados, M. Synovitis: A potential predictive factor of structural progression of medial tibiofemoral knee osteoarthritis - Results of a 1 year longitudinal arthroscopic study in 422 patients. Osteoarthr. Cartil. 2005, 13, 361–367. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef]
- Yang, F.; Zhou, S.; Wang, C.; Huang, Y.; Li, H.; Wang, Y.; Zhu, Z.; Tang, J.; Yan, M. Epigenetic modifications of interleukin-6 in synovial fibroblasts from osteoarthritis patients. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Baker, K.; Grainger, A.; Niu, J.; Clancy, M.; Guermazi, A.; Crema, M.; Hughes, L.; Buckwalter, J.; Wooley, A.; Nevitt, M.; et al. Relation of synovitis to knee pain using contrast-enhanced MRIs. Ann. Rheum. Dis. 2010, 69, 1779–1783. [Google Scholar] [CrossRef]
- Roemer, F.W.; Kassim Javaid, M.; Guermazi, A.; Thomas, M.; Kiran, A.; Keen, R.; King, L.; Arden, N.K. Anatomical distribution of synovitis in knee osteoarthritis and its association with joint effusion assessed on non-enhanced and contrast-enhanced MRI. Osteoarthr. Cartil. 2010, 18, 1269–1274. [Google Scholar] [CrossRef]
- O’Neill, T.W.; Parkes, M.J.; Maricar, N.; Marjanovic, E.J.; Hodgson, R.; Gait, A.D.; Cootes, T.F.; Hutchinson, C.E.; Felson, D.T. Synovial tissue volume: A treatment target in knee osteoarthritis (OA). Ann. Rheum. Dis. 2016, 75, 84–90. [Google Scholar] [CrossRef]
- Hunter, D.J.; McDougall, J.J.; Keefe, F.J. The Symptoms of Osteoarthritis and the Genesis of Pain. Rheum. Dis. Clin. N. Am. 2008, 34, 623–643. [Google Scholar] [CrossRef]
- Fusco, M.; Skaper, S.D.; Coaccioli, S.; Varrassi, G.; Paladini, A. Degenerative Joint Diseases and Neuroinflammation. Pain. Pract. 2017, 17, 522–532. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, M.A.; Conaghan, P.; Le Bars, M.; Baron, G.; Grassi, W.; Martin-Mola, E.; Wakefield, R.; Brasseur, J.L.; So, A.; Backhaus, M.; et al. EULAR report on the use of ultrasonography in painful knee osteoarthritis. Part 1: Prevalence of inflammation in osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Madrid, F.; Karvonen, R.L.; Teitge, R.A.; Miller, P.R.; An, T.; Negendank, W.G. Synovial thickening detected by MR imaging in osteoarthritis of the knee confirmed by biopsy as synovitis. Magn. Reson. Imaging 1995, 13, 177–183. [Google Scholar] [CrossRef]
- Loeuille, D.; Chary-Valckenaere, I.; Champigneulle, J.; Rat, A.C.; Toussaint, F.; Pinzano-Watrin, A.; Goebel, J.C.; Mainard, D.; Blum, A.; Pourel, J.; et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: Correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005, 52, 3492–3501. [Google Scholar] [CrossRef] [PubMed]
- Eymard, F.; Chevalier, X. Inflammation of the infrapatellar fat pad. Jt. Bone Spine 2016, 83, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Paduszyński, W.; Jeśkiewicz, M.; Uchański, P.; Gackowski, S.; Radkowski, M.; Demkow, U. Hoffa’s Fat Pad Abnormality in the Development of Knee Osteoarthritis. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2017; Volume 1039, pp. 95–102. [Google Scholar]
- Roemer, F.W.; Jarraya, M.; Felson, D.T.; Hayashi, D.; Crema, M.D.; Loeuille, D.; Guermazi, A. Magnetic resonance imaging of Hoffa’s fat pad and relevance for osteoarthritis research: A narrative review. Osteoarthr. Cartil. 2016, 24, 383–397. [Google Scholar] [CrossRef]
- Barboza, E.; Hudson, J.; Chang, W.P.; Kovats, S.; Towner, R.A.; Silasi-Mansat, R.; Lupu, F.; Kent, C.; Griffin, T.M. Profibrotic Infrapatellar Fat Pad Remodeling Without M1 Macrophage Polarization Precedes Knee Osteoarthritis in Mice With Diet-Induced Obesity. Arthritis Rheumatol. 2017, 69, 1221–1232. [Google Scholar] [CrossRef]
- Wu, C.L.; Harasymowicz, N.S.; Klimak, M.A.; Collins, K.H.; Guilak, F. The role of macrophages in osteoarthritis and cartilage repair. Osteoarthr. Cartil. 2020, 28, 544–554. [Google Scholar] [CrossRef]
- Ushiyama, T.; Chano, T.; Inoue, K.; Matsusue, Y. Cytokine production in the infrapatellar fat pad: Another source of cytokines in knee synovial fluids. Ann. Rheum. Dis. 2003, 62, 108–112. [Google Scholar] [CrossRef]
- Simopoulou, T.; Malizos, K.N.; Iliopoulos, D.; Stefanou, N.; Papatheodorou, L.; Ioannou, M.; Tsezou, A. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthr. Cartil. 2007, 15, 872–883. [Google Scholar] [CrossRef]
- Englund, M.; Roemer, F.W.; Hayashi, D.; Crema, M.D.; Guermazi, A. Meniscus pathology, osteoarthritis and the treatment controversy. Nat. Rev. Rheumatol. 2012, 8, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Zhang, Y.Q.; Niu, J.B.; Tu, X.; Amin, S.; Clancy, M.; Guermazi, A.; Grigorian, M.; Gale, D.; Felson, D.T. The association of meniscal pathologic changes with cartilage loss in symptomatic knee osteoarthritis. Arthritis Rheum. 2006, 54, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Martel-Pelletier, J.; Pelletier, J.P.; Abram, F.; Raynauld, J.P.; Cicuttini, F.; Jones, G. Meniscal tear as an osteoarthritis risk factor in a largely non-osteoarthritic cohort: A cross-sectional study. J. Rheumatol. 2007, 34, 776–784. [Google Scholar] [PubMed]
- Crema, M.D.; Guermazi, A.; Li, L.; Nogueira-Barbosa, M.H.; Marra, M.D.; Roemer, F.W.; Eckstein, F.; Hellio Le Graverand, M.P.; Wyman, B.T.; Hunter, D.J. The association of prevalent medial meniscal pathology with cartilage loss in the medial tibiofemoral compartment over a 2-year period. Osteoarthr. Cartil. 2010, 18, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Englund, M. The role of biomechanics in the initiation and progression of OA of the knee. Best Pract. Res. Clin. Rheumatol. 2010, 24, 39–46. [Google Scholar] [CrossRef]
- Roemer, F.W.; Guermazi, A.; Hunter, D.J.; Niu, J.; Zhang, Y.; Englund, M.; Javaid, M.K.; Lynch, J.A.; Mohr, A.; Torner, J.; et al. The association of meniscal damage with joint effusion in persons without radiographic osteoarthritis: The Framingham and MOST osteoarthritis studies. Osteoarthr. Cartil. 2009, 17, 748–753. [Google Scholar] [CrossRef]
- Crema, M.D.; Roemer, F.W.; Felson, D.T.; Englund, M.; Wang, K.; Jarraya, M.; Nevitt, M.C.; Marra, M.D.; Torner, J.C.; Lewis, C.E.; et al. Factors associated with meniscal extrusion in knees with or at risk for osteoarthritis: The multicenter osteoarthritis study. Radiology 2012, 264, 494–503. [Google Scholar] [CrossRef]
- Stehling, C.; Souza, R.B.; Le Graverand, M.P.H.; Wyman, B.T.; Li, X.; Majumdar, S.; Link, T.M. Loading of the knee during 3.0 T MRI is associated with significantly increased medial meniscus extrusion in mild and moderate osteoarthritis. Eur. J. Radiol. 2012, 81, 1839–1845. [Google Scholar] [CrossRef]
- van der Voet, J.A.; Runhaar, J.; van der Plas, P.; Vroegindeweij, D.; Oei, E.H.; Bierma-Zeinstra, S.M.A. Baseline meniscal extrusion associated with incident knee osteoarthritis after 30 months in overweight and obese women. Osteoarthr. Cartil. 2017, 25, 1299–1303. [Google Scholar] [CrossRef]
- Liikavainio, T.; Lyytinen, T.; Tyrväinen, E.; Sipilä, S.; Arokoski, J.P. Physical Function and Properties of Quadriceps Femoris Muscle in Men With Knee Osteoarthritis. Arch. Phys. Med. Rehabil. 2008, 89, 2185–2194. [Google Scholar] [CrossRef]
- Alnahdi, A.H.; Zeni, J.A.; Snyder-Mackler, L. Muscle impairments in patients with knee osteoarthritis. Sports Health 2012, 4, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-R.; Yoo, J.; Kim, H. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. [Google Scholar] [CrossRef]
- Roos, E.M.; Herzog, W.; Block, J.A.; Bennell, K.L. Muscle weakness, afferent sensory dysfunction and exercise in knee osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Raynauld, J.-P.; Pelletier, J.-P.; Roubille, C.; Dorais, M.; Abram, F.; Li, W.; Wang, Y.; Fairley, J.; Cicuttini, F.M.; Martel-Pelletier, J. Magnetic Resonance Imaging-Assessed Vastus Medialis Muscle Fat Content and Risk for Knee Osteoarthritis Progression: Relevance From a Clinical Trial. Arthritis Care Res. 2015, 67, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Teichtahl, A.J.; Wluka, A.E.; Wang, Y.; Wijethilake, P.N.; Strauss, B.J.; Proietto, J.; Dixon, J.B.; Jones, G.; Forbes, A.; Cicuttini, F.M. Vastus medialis fat infiltration—A modifiable determinant of knee cartilage loss. Osteoarthr. Cartil. 2015, 23, 2150–2157. [Google Scholar] [CrossRef]
- Krishnasamy, P.; Hall, M.; Robbins, S.R. The role of skeletal muscle in the pathophysiology and management of knee osteoarthritis. Rheumatology 2018, 57, iv22–iv33. [Google Scholar] [CrossRef]
- Rothrauff, B.B.; Jorge, A.; de Sa, D.; Kay, J.; Fu, F.H.; Musahl, V. Anatomic ACL reconstruction reduces risk of post-traumatic osteoarthritis: A systematic review with minimum 10-year follow-up. Knee Surg. Sport Traumatol. Arthrosc. 2019, 28, 1072–1084. [Google Scholar] [CrossRef]
- Wang, Y.; Wluka, A.E.; Berry, P.A.; Siew, T.; Teichtahl, A.J.; Urquhart, D.M.; Lloyd, D.G.; Jones, G.; Cicuttini, F.M. Increase in vastus medialis cross-sectional area is associated with reduced pain, cartilage loss, and joint replacement risk in knee osteoarthritis. Arthritis Rheum. 2012, 64, 3917–3925. [Google Scholar] [CrossRef]
- Mobasheri, A.; Henrotin, Y. Biomarkers of (osteo)arthritis. Biomarkers 2015, 20, 513–518. [Google Scholar] [CrossRef]
- Boehme, K.A.; Rolauffs, B. Onset and Progression of Human Osteoarthritis—Can Growth Factors, Inflammatory Cytokines, or Differential miRNA Expression Concomitantly Induce Proliferation, ECM Degradation, and Inflammation in Articular Cartilage? Int. J. Mol. Sci. 2018, 19, 2282. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell. Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, Ł.A.; Szukiewicz, D. The Role of Inflammatory and Anti-Inflammatory Cytokines in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2014, 2014, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Sharma, A.; Chakraborty, C.; Saibaba, B.; Ahn, M.-E.; Lee, S.-S. Review of Prospects of Biological Fluid Biomarkers in Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 601. [Google Scholar] [CrossRef] [PubMed]
- Mabey, T.; Honsawek, S.; Tanavalee, A.; Yuktanandana, P.; Wilairatana, V.; Poovorawan, Y. Plasma and synovial fluid inflammatory cytokine profiles in primary knee osteoarthritis. Biomarkers 2016, 21, 639–644. [Google Scholar] [CrossRef]
- Zhu, Z.; Otahal, P.; Wang, B.; Jin, X.; Laslett, L.L.L.; Wluka, A.E.E.; Antony, B.; Han, W.; Wang, X.; Winzenberg, T.; et al. Cross-sectional and longitudinal associations between serum inflammatory cytokines and knee bone marrow lesions in patients with knee osteoarthritis. Osteoarthr. Cartil. 2017, 25, 499–505. [Google Scholar] [CrossRef]
- Yang, P.; Tan, J.; Yuan, Z.; Meng, G.; Bi, L.; Liu, J. Expression profile of cytokines and chemokines in osteoarthritis patients: Proinflammatory roles for CXCL8 and CXCL11 to chondrocytes. Int. Immunopharmacol. 2016, 40, 16–23. [Google Scholar] [CrossRef]
- Halász, K.; Kassner, A.; Mörgelin, M.; Heinegård, D. COMP acts as a catalyst in collagen fibrillogenesis. J. Biol. Chem. 2007, 282, 31166–31173. [Google Scholar] [CrossRef]
- Ruan, G.; Xu, J.; Wang, K.; Wu, J.; Zhu, Q.; Ren, J.; Bian, F.; Chang, B.; Bai, X.; Han, W.; et al. Associations between knee structural measures, circulating inflammatory factors and MMP13 in patients with knee osteoarthritis. Osteoarthr. Cartil. 2018, 26, 1063–1069. [Google Scholar] [CrossRef]
- Goldring, M.B.; Berenbaum, F. The Regulation of Chondrocyte Function by Proinflammatory Mediators. Clin. Orthop. Relat. Res. 2004, 427, S37–S46. [Google Scholar] [CrossRef]
- Yang, Q.; Zhou, Y.; Cai, P.; Fu, W.; Wang, J.; Wei, Q.; Li, X. Up-regulated HIF-2α contributes to the Osteoarthritis development through mediating the primary cilia loss. Int. Immunopharmacol. 2019, 75, 105762. [Google Scholar] [CrossRef] [PubMed]
- Alaaeddine, N.; Olee, T.; Hashimoto, S.; Creighton-Achermann, L.; Lotz, M. Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis Rheum. 2001, 44, 1633–1643. [Google Scholar] [CrossRef]
- Akeson, G.; Malemud, C. A Role for Soluble IL-6 Receptor in Osteoarthritis. J. Funct. Morphol. Kinesiol. 2017, 2, 27. [Google Scholar] [CrossRef] [PubMed]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2017, 76, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, W.J.; Tang, S.; Liu, Y.; Wang, J. Correlation between interleukin-6 expression in articular cartilage bone and osteoarthritis. Genet. Mol. Res. 2015, 14, 14189–14195. [Google Scholar] [CrossRef] [PubMed]
- Pearson, M.J.; Herndler-Brandstetter, D.; Tariq, M.A.; Nicholson, T.A.; Philp, A.M.; Smith, H.L.; Davis, E.T.; Jones, S.W.; Lord, J.M. IL-6 secretion in osteoarthritis patients is mediated by chondrocyte-synovial fibroblast cross-talk and is enhanced by obesity. Sci. Rep. 2017, 7, 3451. [Google Scholar] [CrossRef] [PubMed]
- Heinegård, D.; Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.; Petersen, K.K.; Andersen, H.H.; Simonsen, O.; Arendt-Nielsen, L. Serum Inflammatory Markers in Patients with Knee Osteoarthritis: A Proteomic Approach. Clin. J. Pain. 2020, 36, 229–237. [Google Scholar] [CrossRef]
- Larsson, S.; Lohmander, L.S.; Struglics, A. Synovial fluid level of aggrecan ARGS fragments is a more sensitive marker of joint disease than glycosaminoglycan or aggrecan levels: A cross-sectional study. Arthritis Res. Ther. 2009, 11, R92. [Google Scholar] [CrossRef]
- Zhou, J.L.; Fang, H.S.; Peng, H.; Deng, S.; Chen, S.; Li, J.P.; Qiu, B.; Weng, J.Q.; Liu, F. The relationship between HIF-2α and VEGF with radiographic severity in the primary osteoarthritic knee. Yonsei Med. J. 2016, 57, 735–740. [Google Scholar] [CrossRef]
- Venkatesan, J.K.; Rey-Rico, A.; Schmitt, G.; Wezel, A.; Madry, H.; Cucchiarini, M. rAAV-mediated overexpression of TGF-β stably restructures human osteoarthritic articular cartilage in situ. J. Transl. Med. 2013, 11, 211. [Google Scholar] [CrossRef]
- Carballo, C.B.; Coelho, T.R.P.; de Holanda Afonso, R.C.; de O. Faria, J.C.; Alves, T.; Monte, S.M.; Ventura Matioszek, G.M.; Moura-Neto, V.; de Brito, J.M. Osteoarthritic Synovial Fluid and TGF-β1 Induce Interleukin-18 in Articular Chondrocytes. Cartilage 2018, 194760351879614. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Toupet, K.; Maumus, M.; Rozier, P.; Jorgensen, C.; Noël, D. TGFBI secreted by mesenchymal stromal cells ameliorates osteoarthritis and is detected in extracellular vesicles. Biomaterials 2020, 226, 119544. [Google Scholar] [CrossRef]
- Zhen, G.; Wen, C.; Jia, X.; Li, Y.; Crane, J.L.; Mears, S.C.; Askin, F.B.; Frassica, F.J.; Chang, W.; Yao, J.; et al. Inhibition of TGF-β signaling in mesenchymal stem cells of subchondral bone attenuates osteoarthritis. Nat. Med. 2013, 19, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Waly, N.E.; Refaiy, A.; Aborehab, N.M. IL-10 and TGF-β: Roles in chondroprotective effects of Glucosamine in experimental Osteoarthritis? Pathophysiology 2017, 24, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Heard, B.J.; Rosvold, J.M.; Fritzler, M.J.; El-Gabalawy, H.; Wiley, J.P.; Krawetz, R.J. A computational method to differentiate normal individuals, osteoarthritis and rheumatoid arthritis patients using serum biomarkers. J. R. Soc. Interface 2014, 11, 20140428. [Google Scholar] [CrossRef]
- Kisand, K.; Tamm, A.E.; Lintrop, M.; Tamm, A.O. New insights into the natural course of knee osteoarthritis: Early regulation of cytokines and growth factors, with emphasis on sex-dependent angiogenesis and tissue remodeling. A pilot study. Osteoarthr. Cartil. 2018, 26, 1045–1054. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Huebner, J.L.; Haaland, B.; Wong, S.B.S.; Kraus, V.B. Synovial fluid pro-inflammatory profile differs according to the characteristics of knee pain. Osteoarthr. Cartil. 2017, 25, 1420–1427. [Google Scholar] [CrossRef]
- Nees, T.A.; Rosshirt, N.; Zhang, J.A.; Reiner, T.; Sorbi, R.; Tripel, E.; Walker, T.; Schiltenwolf, M.; Hagmann, S.; Moradi, B. Synovial Cytokines Significantly Correlate with Osteoarthritis-Related Knee Pain and Disability: Inflammatory Mediators of Potential Clinical Relevance. J. Clin. Med. 2019, 8, 1343. [Google Scholar] [CrossRef]
- Grieshaber-Bouyer, R.; Kämmerer, T.; Rosshirt, N.; Nees, T.A.; Koniezke, P.; Tripel, E.; Schiltenwolf, M.; Kirsch, J.; Hagmann, S.; Moradi, B. Divergent Mononuclear Cell Participation and Cytokine Release Profiles Define Hip andKnee Osteoarthritis. J. Clin. Med. 2019, 8, 1631. [Google Scholar] [CrossRef]
- Ren, G.; Lutz, I.; Railton, P.; Wiley, J.P.; McAllister, J.; Powell, J.; Krawetz, R.J. Serum and synovial fluid cytokine profiling in hip osteoarthritis: Distinct from knee osteoarthritis and correlated with pain. BMC Musculoskelet. Disord. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- WHO | Obesity. Available online: https://www.who.int/topics/obesity/en/ (accessed on 4 May 2020).
- Blüher, M. Obesity: Global epidemiology and pathogenesis. Nat. Rev. Endocrinol. 2019, 15, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Murphy, L.; Schwartz, T.A.; Helmick, C.G.; Renner, J.B.; Tudor, G.; Koch, G.; Dragomir, A.; Kalsbeek, W.D.; Luta, G.; Jordan, J.M. Lifetime risk of symptomatic knee osteoarthritis. Arthritis Care Res. 2008, 59, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Powell, A. Obesity: A preventable risk factor for large joint osteoarthritis which may act through biomechanical factors. Br. J. Sports Med. 2005, 39, 4–5. [Google Scholar] [CrossRef]
- Aspden, R.M. Obesity punches above its weight in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 65–68. [Google Scholar] [CrossRef]
- Sharma, L.; Song, J.; Felson, D.T.; Cahue, S.; Shamiyeh, E.; Dunlop, D.D. The role of knee alignment in disease progression and functional decline in knee osteoarthritis. J. Am. Med. Assoc. 2001, 286, 188–195. [Google Scholar] [CrossRef]
- Urban, H.; Little, C.B. The role of fat and inflammation in the pathogenesis and management of osteoarthritis. Rheumatology 2018, 57, iv10–iv21. [Google Scholar] [CrossRef]
- Kluzek, S.; Arden, N.K.; Newton, J. Adipokines as potential prognostic biomarkers in patients with acute knee injury. Biomarkers 2015, 20, 519–525. [Google Scholar] [CrossRef]
- Collins, A.T.; Kulvaranon, M.L.; Cutcliffe, H.C.; Utturkar, G.M.; Smith, W.A.R.R.; Spritzer, C.E.; Guilak, F.; DeFrate, L.E. Obesity alters the in vivo mechanical response and biochemical properties of cartilage as measured by MRI. Arthritis Res. Ther. 2018, 20, 232. [Google Scholar] [CrossRef]
- Boyce, L.; Prasad, A.; Barrett, M.; Dawson-Bowling, S.; Millington, S.; Hanna, S.A.; Achan, P. The outcomes of total knee arthroplasty in morbidly obese patients: A systematic review of the literature. Arch. Orthop. Trauma Surg. 2019, 139, 553–560. [Google Scholar] [CrossRef]
- Choi, Y.R.; Collins, K.H.; Lee, J.W.; Kang, H.J.; Guilak, F. Genome Engineering for Osteoarthritis: From Designer Cells to Disease-Modifying Drugs. Tissue Eng. Regen. Med. 2019, 16, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wang, C.; Li, D.; Xu, T.; Myers, J.; Ashton, J.M.; Wang, T.; Zuscik, M.J.; McAlinden, A.; O’Keefe, R.J. DNA methyltransferase 3b regulates articular cartilage homeostasis by altering metabolism. JCI Insight 2017, 2, e93612. [Google Scholar] [CrossRef] [PubMed]
- Cao, K.; Wei, L.; Zhang, Z.; Guo, L.; Zhang, C.; Li, Y.; Sun, C.; Sun, X.; Wang, S.; Li, P.; et al. Decreased histone deacetylase 4 is associated with human osteoarthritis cartilage degeneration by releasing histone deacetylase 4 inhibition of runt-related transcription factor-2 and increasing osteoarthritis-related genes: A novel mechanism of human ost. Arthritis Res. Ther. 2014, 16, 491. [Google Scholar] [CrossRef] [PubMed]
- Richard, D.; Liu, Z.; Cao, J.; Kiapour, A.M.; Willen, J.; Yarlagadda, S.; Jagoda, E.; Kolachalama, V.B.; Sieker, J.T.; Chang, G.H.; et al. Evolutionary Selection and Constraint on Human Knee Chondrocyte Regulation Impacts Osteoarthritis Risk. Cell 2020, 181, 362–381. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Mabuchi, A.; Shi, D.; Kubo, T.; Takatori, Y.; Saito, S.; Fujioka, M.; Sudo, A.; Uchida, A.; Yamamoto, S.; et al. A functional polymorphism in the 5′ UTR of GDF5 is associated with susceptibility to osteoarthritis. Nat. Genet. 2007, 39, 529–533. [Google Scholar] [CrossRef]
- Zengini, E.; Hatzikotoulas, K.; Tachmazidou, I.; Steinberg, J.; Hartwig, F.P.; Southam, L.; Hackinger, S.; Boer, C.G.; Styrkarsdottir, U.; Gilly, A.; et al. Genome-wide analyses using UK Biobank data provide insights into the genetic architecture of osteoarthritis. Nat. Genet. 2018, 50, 549–558. [Google Scholar] [CrossRef]
- Liu, Y.; Chang, J.-C.; Hon, C.-C.; Fukui, N.; Tanaka, N.; Zhang, Z.; Lee, M.T.M.; Minoda, A. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation in osteoarthritis. Sci. Rep. 2018, 8, 15499. [Google Scholar] [CrossRef]
- Yu, X.-M.; Meng, H.-Y.; Yuan, X.-L.; Wang, Y.; Guo, Q.-Y.; Peng, J.; Wang, A.-Y.; Lu, S.-B. MicroRNAs’ Involvement in Osteoarthritis and the Prospects for Treatments. Evid. Based Complement. Altern. Med. 2015, 2015, 1–13. [Google Scholar] [CrossRef]
- Zhang, M.; Lygrissea, K.; Wanga, J. Role of MicroRNA in Osteoarthritis. J. Arthritis 2017, 06, 239. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Zhao, J.; Xu, J.; Geng, Y.; Dai, L.; Huang, Y.; Fu, S.C.; Dai, K.; Zhang, X. MiR-146a facilitates osteoarthritis by regulating cartilage homeostasis via targeting Camk2d and Ppp3r2. Cell Death Dis. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Coutinho De Almeida, R.; Ramos, Y.F.M.; Mahfouz, A.; Den Hollander, W.; Lakenberg, N.; Houtman, E.; Van Hoolwerff, M.; Rodríguez Ruiz, A.; Slagboom, P.E.; Mei, H.; et al. TranslaTional science RNA sequencing data integration reveals an miRNA interactome of osteoarthritis cartilage. Ann. Rheum. Dis. 2019, 78, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Grigelioniene, G.; Suzuki, H.I.; Taylan, F.; Mirzamohammadi, F.; Borochowitz, Z.U.; Ayturk, U.M.; Tzur, S.; Horemuzova, E.; Lindstrand, A.; Weis, M.A.; et al. Gain-of-function mutation of microRNA-140 in human skeletal dysplasia. Nat. Med. 2019, 25, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhao, X.; Mao, G.; Zhang, Z.Z.; Wen, X.; Zhang, C.; Liao, W.; Zhang, Z.Z. MicroRNA-455-3p promotes TGF-β signaling and inhibits osteoarthritis development by directly targeting PAK2. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Hu, G.; Zhao, X.; Wang, C.; Geng, Y.; Zhao, J.; Xu, J.; Zuo, B.; Zhao, C.; Wang, C.; Zhang, X. MicroRNA-145 attenuates TNF-α-driven cartilage matrix degradation in osteoarthritis via direct suppression of MKK4. Cell Death Dis. 2017, 8, e3140. [Google Scholar] [CrossRef]
- Ragni, E.; Perucca Orfei, C.; De Luca, P.; Viganò, M.; Colombini, A.; Lugano, G.; Bollati, V.; de Girolamo, L. miR-22-5p and miR-29a-5p Are Reliable Reference Genes for Analyzing Extracellular Vesicle-Associated miRNAs in Adipose-Derived Mesenchymal Stem Cells and Are Stable under Inflammatory Priming Mimicking Osteoarthritis Condition. Stem Cell Rev. Rep. 2019, 15, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Noël, D.; Pers, Y.M.; Apparailly, F.; Jorgensen, C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat. Rev. Rheumatol. 2016, 12, 211–220. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, L.; Fan, Y.; Liao, L.; Ma, P.X.; Xiao, G.; Chen, D. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Kang, D.; Shin, J.; Cho, Y.; Kim, H.-S.; Gu, Y.-R.; Kim, H.; You, K.T.; Chang, M.J.; Chang, C.B.; Kang, S.-B.; et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 2019, 11, eaar6659. [Google Scholar] [CrossRef]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef]
- Grover, A.K.; Samson, S.E. Benefits of antioxidant supplements for knee osteoarthritis: Rationale and reality. Nutr. J. 2015, 15, 1. [Google Scholar] [CrossRef]
- Blanco, F.J.; Rego, I.; Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Rego-Pérez, I.; Durán-Sotuela, A.; Ramos-Louro, P.; Blanco, F.J. Mitochondrial Genetics and Epigenetics in Osteoarthritis. Front. Genet. 2020, 10, 1335. [Google Scholar] [CrossRef] [PubMed]
- Andia, I.; Maffulli, N. Platelet-rich plasma for managing pain and inflammation in osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 721–730. [Google Scholar] [CrossRef]
- Dohan Ehrenfest, D.M.; Andia, I.; Zumstein, M.A.; Zhang, C.-Q.; Pinto, N.R.; Bielecki, T. Classification of platelet concentrates (Platelet-Rich Plasma-PRP, Platelet-Rich Fibrin-PRF) for topical and infiltrative use in orthopedic and sports medicine: Current consensus, clinical implications and perspectives. Muscle Ligaments Tendons J. 2019, 4, 3–9. [Google Scholar] [CrossRef]
- Wu, Q.; Luo, X.; Xiong, Y.; Liu, G.; Wang, J.; Chen, X.; Mi, B. Platelet-rich plasma versus hyaluronic acid in knee osteoarthritis: A meta-analysis with the consistent ratio of injection. J. Orthop. Surg. 2020, 28, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gato-Calvo, L.; Magalhaes, J.; Ruiz-Romero, C.; Blanco, F.J.; Burguera, E.F. Platelet-rich plasma in osteoarthritis treatment: Review of current evidence. Ther. Adv. Chronic Dis. 2019, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, K.; Vargas-Hernández, J.S.; Sanchez, T.R.; Moreu, N.M.; Mont, M.A.; Higuera, C.A.; Piuzzi, N.S. Are Subchondral Intraosseous Injections Effective and Safe for the Treatment of Knee Osteoarthritis? A Systematic Review. J. Knee Surg. 2019, 32, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Delgado, D.; Pompei, O.; Pérez, J.C.; Sánchez, P.; Garate, A.; Bilbao, A.M.; Fiz, N.; Padilla, S. Treating Severe Knee Osteoarthritis with Combination of Intra-Osseous and Intra-Articular Infiltrations of Platelet-Rich Plasma: An Observational Study. Cartilage 2019, 10, 245–253. [Google Scholar] [CrossRef]
- Ponchel, F.; Burska, A.N.; Hensor, E.M.A.; Raja, R.; Campbell, M.; Emery, P.; Conaghan, P.G. Changes in peripheral blood immune cell composition in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1870–1878. [Google Scholar] [CrossRef]
- Daghestani, H.N.; Kraus, V.B. Inflammatory biomarkers in osteoarthritis. Osteoarthr. Cartil. 2015, 23, 1890–1896. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Medicinal signalling cells: They work, so use them. Nature 2019, 566, 39. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Hariri, R. Body Management: Mesenchymal Stem Cells Control the Internal Regenerator. Stem Cells Transl. Med. 2015, 4, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. What’s in a Name? Tissue Eng. Part A 2010, 16, 2415–2417. [Google Scholar] [CrossRef] [PubMed]
- McGonagle, D.; Baboolal, T.G.; Jones, E. Native joint-resident mesenchymal stem cells for cartilage repair in osteoarthritis. Nat. Rev. Rheumatol. 2017, 13, 719–730. [Google Scholar] [CrossRef] [PubMed]
- De Windt, T.S.; Saris, D.B.F.; Slaper-Cortenbach, I.C.M.; Van Rijen, M.H.P.; Gawlitta, D.; Creemers, L.B.; De Weger, R.A.; Dhert, W.J.A.; Vonk, L.A. Direct cell-cell contact with chondrocytes is a key mechanism in multipotent mesenchymal stromal cell-mediated chondrogenesis. Tissue Eng. Part A 2015, 21, 2536–2547. [Google Scholar] [CrossRef]
- Mancuso, P.; Raman, S.; Glynn, A.; Barry, F.; Murphy, J.M. Mesenchymal Stem Cell Therapy for Osteoarthritis: The Critical Role of the Cell Secretome. Front. Bioeng. Biotechnol. 2019, 7, 9. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Discher, D.E.; Péault, B.M.; Phinney, D.G.; Hare, J.M.; Caplan, A.I. Mesenchymal stem cell perspective: Cell biology to clinical progress. Npj Regen. Med. 2019, 4, 22. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal Stem Cell-Mediated Immunosuppression Occurs via Concerted Action of Chemokines and Nitric Oxide. Cell Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, D.; Liu, Z.; Zhou, F.; Dai, J.; Wu, B.; Zhou, J.; Heng, B.C.; Zou, X.H.; Ouyang, H.; et al. Exosomes from embryonic mesenchymal stem cells alleviate osteoarthritis through balancing synthesis and degradation of cartilage extracellular matrix. Stem Cell Res. Ther. 2017, 8, 189. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Ha, C.-W.; Park, Y.-B.; Nam, E.; Lee, J.-E.; Lee, H.-J. Intra-articular injection of mesenchymal stem cells for clinical outcomes and cartilage repair in osteoarthritis of the knee: A meta-analysis of randomized controlled trials. Arch. Orthop. Trauma Surg. 2019, 139, 971–980. [Google Scholar] [CrossRef]
- Caplan, A.I.; Correa, D. The MSC: An Injury Drugstore. Cell Stem Cell 2011, 9, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef]
- Hassan, H.T.; El-Sheemy, M. Adult bone-marrow stem cells and their potential in medicine. J. R. Soc. Med. 2004, 97, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Chahal, J.; Gómez-Aristizábal, A.; Shestopaloff, K.; Bhatt, S.; Chaboureau, A.; Fazio, A.; Chisholm, J.; Weston, A.; Chiovitti, J.; Keating, A.; et al. Bone Marrow Mesenchymal Stromal Cell Treatment in Patients with Osteoarthritis Results in Overall Improvement in Pain and Symptoms and Reduces Synovial Inflammation. Stem Cells Transl. Med. 2019, 8, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Chullikana, A.; Rengasamy, M.; Shetty, N.; Pandey, V.; Agarwal, V.; Wagh, S.Y.; Vellotare, P.K.; Damodaran, D.; Viswanathan, P.; et al. Efficacy and safety of adult human bone marrow-derived, cultured, pooled, allogeneic mesenchymal stromal cells (Stempeucel®): Preclinical and clinical trial in osteoarthritis of the knee joint. Arthritis Res. Ther. 2016, 18, 301. [Google Scholar] [CrossRef]
- Vega, A.; Martín-Ferrero, M.A.; Del Canto, F.; Alberca, M.; García, V.; Munar, A.; Orozco, L.; Soler, R.; Fuertes, J.J.; Huguet, M.; et al. Treatment of knee osteoarthritis with allogeneic bone marrow mesenchymal stem cells: A randomized controlled trial. Transplantation 2015, 99, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.E.; Hussein, K.A.; Helwa, I.; Abdelsamid, M.F.; Aguilar-Perez, A.; Mohsen, I.; Hunter, M.; Hamrick, M.W.; Isales, C.M.; Elsalanty, M.; et al. Meta-Analysis and Evidence Base for the Efficacy of Autologous Bone Marrow Mesenchymal Stem Cells in Knee Cartilage Repair: Methodological Guidelines and Quality Assessment. Stem Cells Int. 2019, 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Maumus, M.; Pers, Y.-M.; Ruiz, M.; Jorgensen, C.; Noël, D. Cellules souches mésenchymateuses et médecine régénératrice. Méd. Sci. 2018, 34, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Hudetz, D.; Jeleč, Ž.; Rod, E.; Borić, I.; Plečko, M.; Primorac, D. The Future of Cartilage Repair. In Personalized Medicine in Healthcare Systems: Legal, Medical and Economic Implications; Bodiroga-Vukobrat, N., Rukavina, D., Pavelić, K., Sander, G.G., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 375–411. ISBN 978-3-030-16465-2. [Google Scholar]
- Hudetz, D.; Borić, I.; Rod, E.; Jeleč, Ž.; Radić, A.; Vrdoljak, T.; Skelin, A.; Lauc, G.; Trbojević-Akmačić, I.; Plečko, M.; et al. The Effect of Intra-articular Injection of Autologous Microfragmented Fat Tissue on Proteoglycan Synthesis in Patients with Knee Osteoarthritis. Genes 2017, 8, 270. [Google Scholar] [CrossRef] [PubMed]
- Hudetz, D.; Borić, I.; Rod, E.; Jeleč, Ž.; Kunovac, B.; Polašek, O.; Vrdoljak, T.; Plečko, M.; Skelin, A.; Polančec, D.; et al. Early results of intra-articular micro-fragmented lipoaspirate treatment in patients with late stages knee osteoarthritis: A prospective study. Croat. Med. J. 2019, 60, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Polancec, D.; Zenic, L.; Hudetz, D.; Boric, I.; Jelec, Z.; Rod, E.; Vrdoljak, T.; Skelin, A.; Plecko, M.; Turkalj, M.; et al. Immunophenotyping of a Stromal Vascular Fraction from Microfragmented Lipoaspirate Used in Osteoarthritis Cartilage Treatment and Its Lipoaspirate Counterpart. Genes 2019, 10, 474. [Google Scholar] [CrossRef]
- Borić, I.; Hudetz, D.; Rod, E.; Jeleč, Ž.; Vrdoljak, T.; Skelin, A.; Polašek, O.; Plečko, M.; Trbojević-Akmačić, I.; Lauc, G.; et al. A 24-Month Follow-Up Study of the Effect of Intra-Articular Injection of Autologous Microfragmented Fat Tissue on Proteoglycan Synthesis in Patients with Knee Osteoarthritis. Genes 2019, 10, 1051. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Screpis, D.; Di Donato, S.L.; Bonetti, S.; Piovan, G.; Zorzi, C. Autologous micro-fragmented adipose tissue for the treatment of diffuse degenerative knee osteoarthritis: An update at 3 year follow-up. J. Exp. Orthop. 2018, 5, 52. [Google Scholar] [CrossRef] [PubMed]
- Mautner, K.; Bowers, R.; Easley, K.; Fausel, Z.; Robinson, R. Functional Outcomes Following Microfragmented Adipose Tissue Versus Bone Marrow Aspirate Concentrate Injections for Symptomatic Knee Osteoarthritis. Stem Cells Transl. Med. 2019, 8, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Russo, A.; Condello, V.; Madonna, V.; Guerriero, M.; Zorzi, C. Autologous and micro-fragmented adipose tissue for the treatment of diffuse degenerative knee osteoarthritis. J. Exp. Orthop. 2017, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Peretti, G.M.; Ulivi, M.; De Girolamo, L.; Meroni, V.; Lombardo, M.D.; Mangiavini, L. Evaluation of the use of autologous micro-fragmented adipose tissue in the treatment of knee osteoarthritis: Preliminary results of a randomized controlled trial. J. Biol. Regul. Homeost. Agents 2018, 32, 193–199. [Google Scholar]
- Freitag, J.; Bates, D.; Wickham, J.; Shah, K.; Huguenin, L.; Tenen, A.; Paterson, K.; Boyd, R. Adipose-derived mesenchymal stem cell therapy in the treatment of knee osteoarthritis: A randomized controlled trial. Regen. Med. 2019, 14, 213–230. [Google Scholar] [CrossRef]
- Yun, S.; Ku, S.-K.; Kwon, Y.-S. Adipose-derived mesenchymal stem cells and platelet-rich plasma synergistically ameliorate the surgical-induced osteoarthritis in Beagle dogs. J. Orthop. Surg. Res. 2016, 11, 9. [Google Scholar] [CrossRef]
- Pak, J.; Chang, J.-J.; Lee, J.H.; Lee, S.H. Safety reporting on implantation of autologous adipose tissue-derived stem cells with platelet-rich plasma into human articular joints. BMC Musculoskelet. Disord. 2013, 14, 337. [Google Scholar] [CrossRef]
- Jayaram, P.; Ikpeama, U.; Rothenberg, J.B.; Malanga, G.A. Bone Marrow-Derived and Adipose-Derived Mesenchymal Stem Cell Therapy in Primary Knee Osteoarthritis: A Narrative Review. PM&R 2019, 11, 177–191. [Google Scholar] [CrossRef]
- Shariatzadeh, M.; Song, J.; Wilson, S.L. The efficacy of different sources of mesenchymal stem cells for the treatment of knee osteoarthritis. Cell Tissue Res. 2019, 378, 399–410. [Google Scholar] [CrossRef]
- Wu, X.; Wang, Y.; Xiao, Y.; Crawford, R.; Mao, X.; Prasadam, I. Extracellular vesicles: Potential role in osteoarthritis regenerative medicine. J. Orthop. Transl. 2020, 21, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Lener, T.; Gimona, M.; Aigner, L.; Börger, V.; Buzas, E.; Camussi, G.; Chaput, N.; Chatterjee, D.; Court, F.A.; del Portillo, H.A.; et al. Applying extracellular vesicles based therapeutics in clinical trials—An ISEV position paper. J. Extracell. Vesicles 2015, 4, 30087. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.J.; Jackson, M.V.; Cunningham, E.K.; Kissenpfennig, A.; McAuley, D.F.; O’Kane, C.M.; Krasnodembskaya, A.D. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am. J. Respir. Crit. Care Med. 2017, 196, 1275–1286. [Google Scholar] [CrossRef]
- Tan, S.S.H.; Tjio, C.K.E.; Wong, J.R.Y.; Wong, K.L.; Chew, J.R.J.; Hui, J.H.P.; Toh, W.S. Mesenchymal Stem Cell Exosomes for Cartilage Regeneration: A Systematic Review of Preclinical In Vivo Studies. Tissue Eng. Part B Rev. 2020, 2019, 0326. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, Y.; Yang, G.; Chen, X.; Zhang, Y.; Cao, G.; Wang, J.; Sun, Y.; Zhang, P.; Fan, M.; et al. Transforming Growth Factor-Beta-Regulated miR-24 Promotes Skeletal Muscle Differentiation. Nucleic Acids Res. 2008, 36, 2690–2699. [Google Scholar] [CrossRef]
- Fleury, A.; Martinez, M.C.; Le Lay, S. Extracellular Vesicles as Therapeutic Tools in Cardiovascular Diseases. Front. Immunol. 2014, 5, 370. [Google Scholar] [CrossRef]
- Goldie, B.J.; Dun, M.D.; Lin, M.; Smith, N.D.; Verrills, N.M.; Dayas, C.V.; Cairns, M.J. Activity-associated miRNA are packaged in Map1b-enriched exosomes released from depolarized neurons. Nucleic Acids Res. 2014, 42, 9195–9208. [Google Scholar] [CrossRef]
- Dell’Isola, A.; Allan, R.; Smith, S.L.; Marreiros, S.S.P.; Steultjens, M. Identification of clinical phenotypes in knee osteoarthritis: A systematic review of the literature. BMC Musculoskelet. Disord. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Dell’Isola, A.; Steultjens, M.; Dell’Isola, A.; Steultjens, M.; Isola, A.D.; Steultjens, M. Classification of patients with knee osteoarthritis in clinical phenotypes: Data from the osteoarthritis initiative. PLoS ONE 2018, 13, e191045. [Google Scholar] [CrossRef] [PubMed]
- Van Spil, W.E.; Kubassova, O.; Boesen, M.; Bay-Jensen, A.-C.; Mobasheri, A. Osteoarthritis phenotypes and novel therapeutic targets. Biochem. Pharmacol. 2019, 165, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-M.; Chen, Y.-H.; Sun, H.; Tsai, S.-J. Fibroblast growth factors: Potential novel targets for regenerative therapy of osteoarthritis. Chin. J. Physiol. 2019, 62, 2. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.; Farran, A.; Mohanraj, B.; Guehring, H.; Cocca, R.; Rabut, E.; Mauck, R.; Dodge, G. Recombinant human FGF18 preserves depth-dependent mechanical inhomogeneity in articular cartilage. Eur. Cells Mater. 2019, 38, 23–38. [Google Scholar] [CrossRef]
- Eckstein, F.; Kraines, J.L.; Aydemir, A.; Wirth, W.; Maschek, S.; Hochberg, M.C. Intra-articular sprifermin reduces cartilage loss in addition to increasing cartilage gain independent of location in the femorotibial joint: Post-hoc analysis of a randomised, placebo-controlled phase II clinical trial. Ann. Rheum. Dis. 2020, 79, 525–528. [Google Scholar] [CrossRef]
- Hochberg, M.C.; Guermazi, A.; Guehring, H.; Aydemir, A.; Wax, S.; Fleuranceau-Morel, P.; Reinstrup Bihlet, A.; Byrjalsen, I.; Ragnar Andersen, J.; Eckstein, F. Effect of Intra-Articular Sprifermin vs Placebo on Femorotibial Joint Cartilage Thickness in Patients With Osteoarthritis. JAMA 2019, 322, 1360. [Google Scholar] [CrossRef]
- Gigout, A.; Guehring, H.; Froemel, D.; Meurer, A.; Ladel, C.; Reker, D.; Bay-Jensen, A.C.; Karsdal, M.A.; Lindemann, S. Sprifermin (rhFGF18) enables proliferation of chondrocytes producing a hyaline cartilage matrix. Osteoarthr. Cartil. 2017, 25, 1858–1867. [Google Scholar] [CrossRef]
- Gregori, D.; Giacovelli, G.; Minto, C.; Barbetta, B.; Gualtieri, F.; Azzolina, D.; Vaghi, P.; Rovati, L.C. Association of Pharmacological Treatments With Long-term Pain Control in Patients With Knee Osteoarthritis. JAMA 2018, 320, 2564. [Google Scholar] [CrossRef]
- Stöve, J.; Schneider-Wald, B.; Scharf, H.P.; Schwarz, M.L. Bone morphogenetic protein 7 (bmp-7) stimulates Proteoglycan synthesis in human osteoarthritic chondrocytes in vitro. Biomed. Pharmacother. 2006, 60, 639–643. [Google Scholar] [CrossRef]
- Bone morphogenetic protein 7 inhibits cartilage degradation in a rabbit model of osteoarthritis. Nat. Clin. Pract. Rheumatol. 2009, 5, 4. [CrossRef]
- Hayashi, M.; Muneta, T.; Ju, Y.-J.; Mochizuki, T.; Sekiya, I. Weekly intra-articular injections of bone morphogenetic protein-7 inhibits osteoarthritis progression. Arthritis Res. Ther. 2008, 10, R118. [Google Scholar] [CrossRef] [PubMed]
- Hunter, D.J.; Pike, M.C.; Jonas, B.L.; Kissin, E.; Krop, J.; McAlindon, T. Phase 1 safety and tolerability study of BMP-7 in symptomatic knee osteoarthritis. BMC Musculoskelet. Disord. 2010, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Mimpen, J.Y.; Snelling, S.J.B. Chondroprotective Factors in Osteoarthritis: A Joint Affair. Curr. Rheumatol. Rep. 2019, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Oo, W.M.; Yu, S.P.C.; Daniel, M.S.; Hunter, D.J. Disease-modifying drugs in osteoarthritis: Current understanding and future therapeutics. Expert Opin. Emerg. Drugs 2018, 23, 331–347. [Google Scholar] [CrossRef]
- Shepard, H.M.; Phillips, G.L.; Thanos, C.D.; Feldmann, M. Developments in therapy with monoclonal antibodies and related proteins. Clin. Med. J. R. Coll. Physicians Lond. 2017, 17, 220–232. [Google Scholar] [CrossRef]
- Das, V.; Kc, R.; Li, X.; O-Sullivan, I.S.; van Wijnen, A.J.; Kroin, J.S.; Pytowski, B.; Applegate, D.T.; Votta-Velis, G.; Ripper, R.L.; et al. Blockade of vascular endothelial growth factor receptor-1 (Flt-1), reveals a novel analgesic for osteoarthritis-induced joint pain. Gene Rep. 2018, 11, 94–100. [Google Scholar] [CrossRef]
- Kan, S.-L.; Li, Y.; Ning, G.-Z.; Yuan, Z.-F.; Chen, L.-X.; Bi, M.-C.; Sun, J.-C.; Feng, S.-Q. Tanezumab for Patients with Osteoarthritis of the Knee: A Meta-Analysis. PLoS ONE 2016, 11, e0157105. [Google Scholar] [CrossRef]
- Schnitzer, T.J.; Easton, R.; Pang, S.; Levinson, D.J.; Pixton, G.; Viktrup, L.; Davignon, I.; Brown, M.T.; West, C.R.; Verburg, K.M. Effect of Tanezumab on Joint Pain, Physical Function, and Patient Global Assessment of Osteoarthritis among Patients with Osteoarthritis of the Hip or Knee: A Randomized Clinical Trial. JAMA J. Am. Med. Assoc. 2019, 322, 37–48. [Google Scholar] [CrossRef]
- Tiseo, P.J.; Kivitz, A.J.; Ervin, J.E.; Ren, H.; Mellis, S.J. Fasinumab (REGN475), an antibody against nerve growth factor for the treatment of pain: Results from a double-blind, placebo-controlled exploratory study in osteoarthritis of the knee. Pain 2014, 155, 1245–1252. [Google Scholar] [CrossRef]
- Gow, J.M.; Tsuji, W.H.; Williams, G.J.; Mytych, D.; Sciberras, D.; Searle, S.L.; Mant, T.; Gibbs, J.P. Safety, tolerability, pharmacokinetics, and efficacy of AMG 403, a human anti-nerve growth factor monoclonal antibody, in two phase I studies with healthy volunteers and knee osteoarthritis subjects. Arthritis Res. Ther. 2015, 17, 282. [Google Scholar] [CrossRef]
- Miller, R.E.; Tran, P.B.; Ishihara, S.; Larkin, J.; Malfait, A.M. Therapeutic effects of an anti-ADAMTS-5 antibody on joint damage and mechanical allodynia in a murine model of osteoarthritis. Osteoarthr. Cartil. 2016, 24, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Nixon, A.J.; Grol, M.W.; Lang, H.M.; Ruan, M.Z.C.; Stone, A.; Begum, L.; Chen, Y.; Dawson, B.; Gannon, F.; Plutizki, S.; et al. Disease-Modifying Osteoarthritis Treatment With Interleukin-1 Receptor Antagonist Gene Therapy in Small and Large Animal Models. Arthritis Rheumatol. 2018, 70, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next generation of CRISPR–Cas technologies and applications. Nat. Rev. Mol. Cell Biol. 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Huang, J.; Fan, Y.; Li, J.; You, T.; He, S.; Xiao, G.; Chen, D. Exploration of CRISPR/Cas9-based gene editing as therapy for osteoarthritis. Ann. Rheum. Dis. 2019, 78, 676–682. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primorac, D.; Molnar, V.; Rod, E.; Jeleč, Ž.; Čukelj, F.; Matišić, V.; Vrdoljak, T.; Hudetz, D.; Hajsok, H.; Borić, I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes 2020, 11, 854. https://doi.org/10.3390/genes11080854
Primorac D, Molnar V, Rod E, Jeleč Ž, Čukelj F, Matišić V, Vrdoljak T, Hudetz D, Hajsok H, Borić I. Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes. 2020; 11(8):854. https://doi.org/10.3390/genes11080854
Chicago/Turabian StylePrimorac, Dragan, Vilim Molnar, Eduard Rod, Željko Jeleč, Fabijan Čukelj, Vid Matišić, Trpimir Vrdoljak, Damir Hudetz, Hana Hajsok, and Igor Borić. 2020. "Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations" Genes 11, no. 8: 854. https://doi.org/10.3390/genes11080854
APA StylePrimorac, D., Molnar, V., Rod, E., Jeleč, Ž., Čukelj, F., Matišić, V., Vrdoljak, T., Hudetz, D., Hajsok, H., & Borić, I. (2020). Knee Osteoarthritis: A Review of Pathogenesis and State-Of-The-Art Non-Operative Therapeutic Considerations. Genes, 11(8), 854. https://doi.org/10.3390/genes11080854