Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report

 ,
,  ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Case Report
2.2. Molecular Genetic Studies
2.2.1. NGS Studies
2.2.2. Confirmation and Segregation Analysis
2.3. Cultured Skin Fibroblasts and Cell Growth Rate
2.4. Mitochondrial Respiratory Chain Activities in Fibroblasts
2.5. Mitochondrial Respiration Assays in Fibroblasts
2.6. mRNA Analysis Cultured Fibroblasts
2.7. Western Blot: Assessment of NDUF13 Protein Steady-State Levels in Cultured Fibroblast
2.8. Mitochondrial Supercomplexes Analysis and Complex I In-Gel Activity Assay
2.9. Statistical Analisis
3. Results
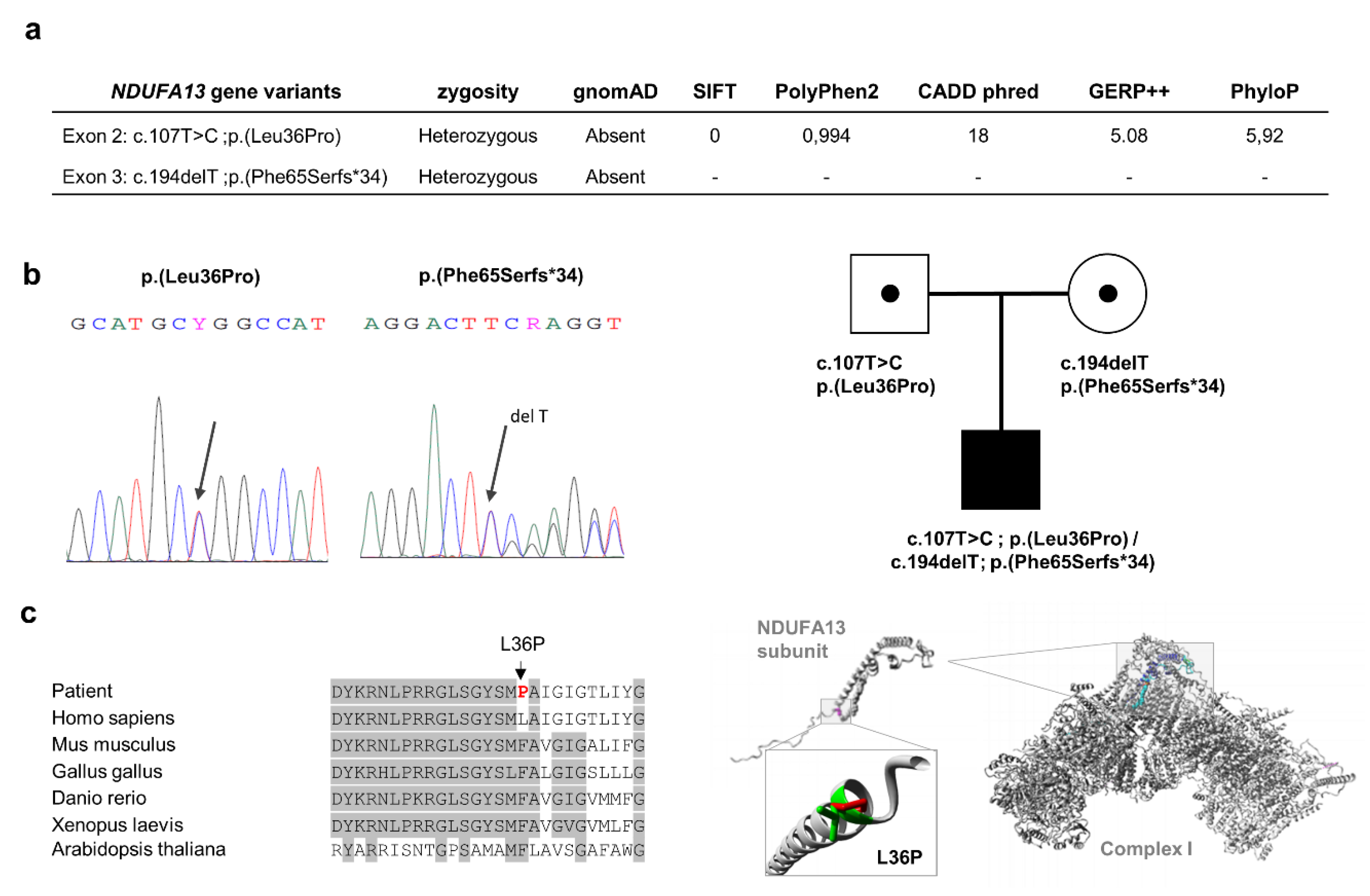
3.1. Molecular Genetics
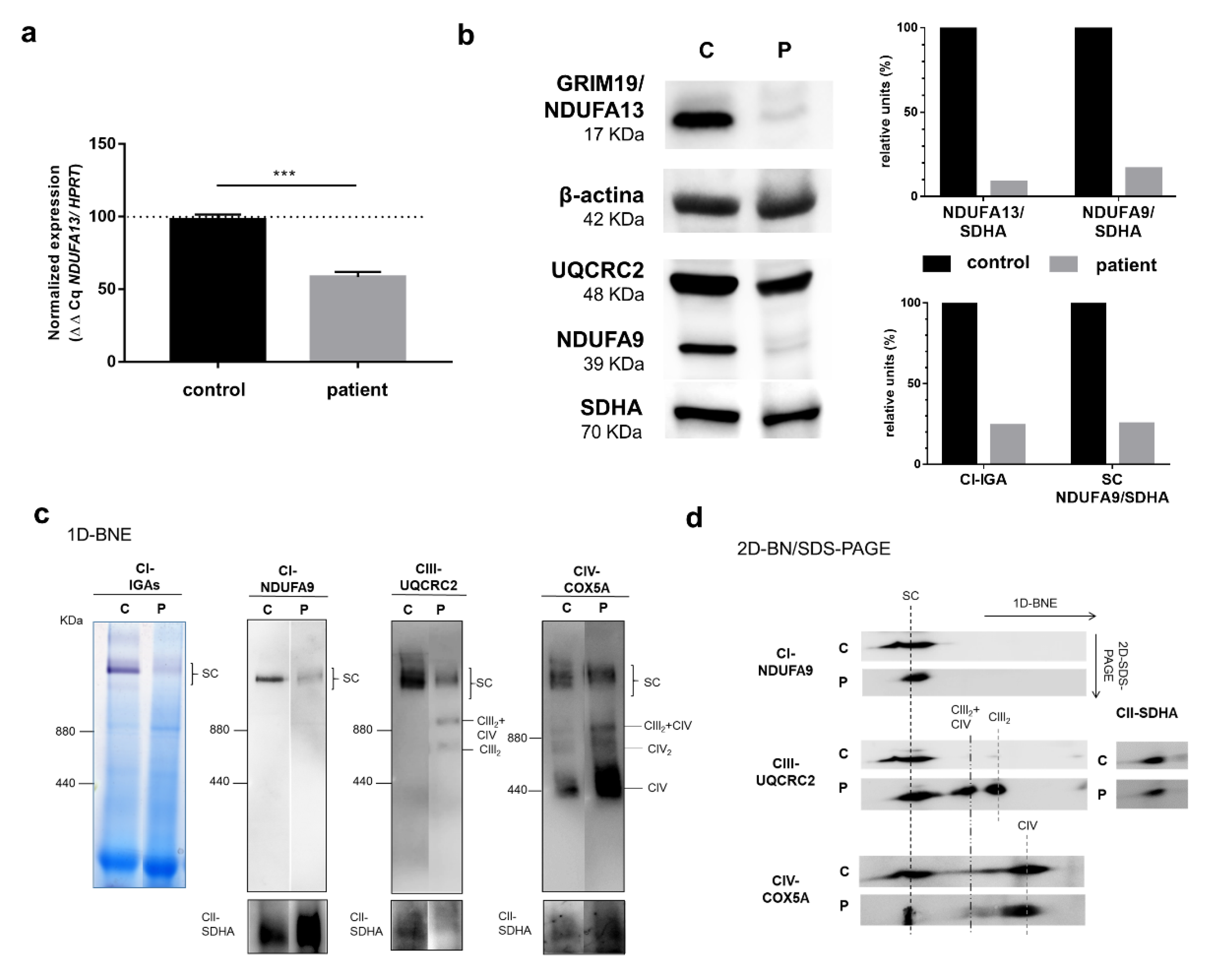
3.2. Biochemical and Cellular Studies in Cultured Skin Fibroblasts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wirth, C.; Brandt, U.; Hunte, C.; Zickermann, V. Structure and function of mitochondrial complex I. Biochim. Biophys. Acta (BBA) Bioenerg. 2016, 1857, 902–914. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nature 2014, 515, 80–84. [Google Scholar] [CrossRef]
- Bugiani, M.; Invernizzi, F.; Alberio, S.; Briem, E.; Lamantea, E.; Carrara, F.; Moroni, I.; Farina, L.; Spada, M.; Donati, M.; et al. Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta (BBA) Gen. Subj. 2004, 1659, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Fassone, E.; Rahman, S. Complex I deficiency: Clinical features, biochemistry and molecular genetics. J. Med. Genet. 2012, 49, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Björkman, K.; Sofou, K.; Darin, N.; Holme, E.; Kollberg, G.; Asin-Cayuela, J.; Dahle, K.M.H.; Oldfors, A.; Moslemi, A.-R.; Tulinius, M. Broad phenotypic variability in patients with complex I deficiency due to mutations in NDUFS1 and NDUFV1. Mitochondrion 2015, 21, 33–40. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Emmanuele, V. The Clinical Spectrum of Nuclear DNA-Related Mitochondrial Disorders. In Mitochondrial Disorders Caused by Nuclear Genes; Wong, L.-J.C., Ed.; Springer: New York, NY, USA, 2013; pp. 3–25. [Google Scholar]
- Leigh, D. Subacute necrotizing encephalomyelopathy in an infant. J. Neurol. Neurosurg. Psychiatr. 1951, 14, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Kim, H.; Lim, B.C.; Hwang, H.; Choi, J.; Kim, K.J.; Hwang, Y.S.; Chae, J.-H. Leigh Syndrome in Childhood: Neurologic Progression and Functional Outcome. J. Clin. Neurol. 2016, 12, 181–187. [Google Scholar] [CrossRef]
- Bonfante, E.; Koenig, M.K.; Adejumo, R.B.; Perinjelil, V.; Riascos, R. The neuroimaging of Leigh syndrome: Case series and review of the literature. Pediatr. Radiol. 2016, 46, 443–451. [Google Scholar] [CrossRef]
- Rahman, S.; Blok, R.B.; Dahl, H.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2015, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Legati, A.; Reyes, A.; Nasca, A.; Invernizzi, F.; Lamantea, E.; Tiranti, V.; Garavaglia, B.; Lamperti, C.; Ardissone, A.; Moroni, I.; et al. New genes and pathomechanisms in mitochondrial disorders unraveled by NGS technologies. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1857, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; He, F.; Yin, F.; Chen, C.; Wu, L.; Yang, L.; Peng, J. The use of targeted genomic capture and massively parallel sequencing in diagnosis of Chinese Leukoencephalopathies. Sci. Rep. 2016, 6, 35936. [Google Scholar] [CrossRef]
- Rubegni, A.; Pisano, T.; Bacci, G.; Tessa, A.; Battini, R.; Procopio, E.; Giglio, S.; Pasquariello, R.; Santorelli, F.M.; Guerrini, R.; et al. Leigh-like neuroimaging features associated with new biallelic mutations in OPA1. Eur. J. Paediatr. Neurol. 2017, 21, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Compton, A.G.; Hershman, S.; Lim, S.C.; Lieber, D.S.; Tucker, E.J.; Laskowski, A.; Garone, C.; Liu, S.; Jaffe, D.B.; et al. Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing. Sci. Transl. Med. 2012, 4, 118ra10. [Google Scholar] [CrossRef] [PubMed]
- Angebault, C.; Charif, M.; Guegen, N.; Piro-Megy, C.; De Camaret, B.M.; Procaccio, V.; Guichet, P.-O.; Hebrard, M.; Manes, G.; Leboucq, N.; et al. Mutation in NDUFA13/GRIM19 leads to early onset hypotonia, dyskinesia and sensorial deficiencies, and mitochondrial complex I instability. Hum. Mol. Genet. 2015, 24, 3948–3955. [Google Scholar] [CrossRef]
- Álvarez-Iglesias, V.; Barros, F.; Carracedo, Á.; Salas, A. Minisequencing mitochondrial DNA pathogenic mutations. BMC Med. Genet. 2008, 9, 26. [Google Scholar] [CrossRef]
- Weissensteiner, H.; Pacher, D.; Kloss-Brandstätter, A.; Forer, L.; Specht, G.; Bandelt, H.-J.; Kronenberg, F.; Salas, A.; Schönherr, S. HaploGrep 2: Mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res. 2016, 44, W58–W63. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Medja, F.; Allouche, S.; Frachon, P.; Jardel, C.; Malgat, M.; De Camaret, B.M.; Slama, A.; Lunardi, J.; Mazat, J.; Lombès, A. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009, 9, 331–339. [Google Scholar] [CrossRef]
- Moran, M.; Rivera, H.; Sánchez-Aragó, M.; Blázquez, A.; Merinero, B.; Ugalde, C.; Arenas, J.; Cuezva, J.; Martin, M.A. Mitochondrial bioenergetics and dynamics interplay in complex I-deficient fibroblasts. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 443–453. [Google Scholar] [CrossRef]
- Desai, R.; Frazier, A.E.; Durigon, R.; Patel, H.; Jones, A.W.; Rosa, I.D.; Lake, N.J.; Compton, A.G.; Mountford, H.; Tucker, E.J.; et al. ATAD3 gene cluster deletions cause cerebellar dysfunction associated with altered mitochondrial DNA and cholesterol metabolism. Brain 2017, 140, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.J. Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 2002, 26, 327–334. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Venselaar, H.; Beek, T.A.T.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinforma. 2010, 11, 548. [Google Scholar] [CrossRef]
- Gray, T.M.; Arnoys, E.J.; Blankespoor, S.; Born, T.; Jagar, R.; Everman, R.; Plowman, D.; Stair, A.; Zhang, D. Destabilizing effect of proline substitutions in two helical regions of T4 lysozyme: Leucine 66 to proline and leucine 91 to proline. Protein Sci. 2008, 5, 742–751. [Google Scholar] [CrossRef]
- Saada, A. Mitochondria: Mitochondrial OXPHOS (dys) function ex vivo—The use of primary fibroblasts. Int. J. Biochem. Cell Boil. 2014, 48, 60–65. [Google Scholar] [CrossRef]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L.G. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef]
- Angell, J.E.; Lindner, D.J.; Shapiro, P.S.; Hofmann, E.R.; Kalvakolanu, D.V. Identification of GRIM-19, a Novel Cell Death-regulatory Gene Induced by the Interferon-β and Retinoic Acid Combination, Using a Genetic Approach. J. Boil. Chem. 2000, 275, 33416–33426. [Google Scholar] [CrossRef]
- Fearnley, I.M.; Carroll, J.; Shannon, R.J.; Runswick, M.J.; Walker, J.E.; Hirst, J. GRIM-19, a Cell Death Regulatory Gene Product, Is a Subunit of Bovine Mitochondrial NADH:Ubiquinone Oxidoreductase (Complex I). J. Boil. Chem. 2001, 276, 38345–38348. [Google Scholar] [CrossRef]
- Moreira, S.; Correia, M.; Soares, P.; Máximo, V. GRIM-19 function in cancer development. Mitochondrion 2011, 11, 693–699. [Google Scholar] [CrossRef]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III -mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-López, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 1–20. [Google Scholar] [CrossRef]
- Wu, M.; Gu, J.; Guo, R.; Huang, Y.; Yang, M. Structure of Mammalian Respiratory Supercomplex I 1 III 2 IV 1. Cell 2016, 167, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; Rodenburg, R.J.T.; Van Der Knaap, M.S.; Willemsen, M.A.A.P.; Sperl, W.; Laugel, V.; Östergaard, E.; Tarnopolsky, M.; Martin, M.A.; Nesbitt, V.; et al. Natural disease course and genotype-phenotype correlations in Complex I deficiency caused by nuclear gene defects: What we learned from 130 cases. J. Inherit. Metab. Dis. 2012, 35, 737–747. [Google Scholar] [CrossRef]
- Rodenburg, R.J.T. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1857, 938–945. [Google Scholar] [CrossRef]
- Witters, P.; Saada, A.; Honzik, T.; Tesarova, M.; Kleinle, S.; Horvath, R.; Goldstein, A.; Morava, E. Revisiting mitochondrial diagnostic criteria in the new era of genomics. Genet. Med. 2017, 20, 444–451. [Google Scholar] [CrossRef] [PubMed]
- Baldo, M.S.; Vilarinho, L.; Schubert, M.B. Molecular basis of Leigh syndrome: A current look. Orphanet J. Rare Dis. 2020, 15, 31. [Google Scholar] [CrossRef]
- Stroud, D.A.; Surgenor, E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Skeletal Muscle | Fibroblasts | |||
|---|---|---|---|---|
| Patient (Controls) 1 | % 2 | Patient (Controls) 3 | % 4 | |
| CI (NADH–DB oxidoreductase) | 8.1 (11.3–24.7) | 72% | 7.1 (20.4 ± 2.3) | 35% |
| CII (Succinate dehydrogenase) | 4.7 (5.8–19.9) | 81% | 58.5 (48.4 ± 7.16) | 120% |
| CIII (DBH2–Cytochrome c oxidoreductase) | 52.3 (31–127) | 169% | 162 (160 ± 22) | 101% |
| CIV (Cytochrome c oxidase) | 21.1 (20–79.2) | 106% | 66.2 (55.4 ± 14.3) | 119% |
| CS (Citrate synthase) 5 | 212 (102–257) | - | 47.1 (77.0 ± 27.0) | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Quintana, A.; García-Consuegra, I.; Belanger-Quintana, A.; Serrano-Lorenzo, P.; Lucia, A.; Blázquez, A.; Docampo, J.; Ugalde, C.; Morán, M.; Arenas, J.; et al. Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report. Genes 2020, 11, 855. https://doi.org/10.3390/genes11080855
González-Quintana A, García-Consuegra I, Belanger-Quintana A, Serrano-Lorenzo P, Lucia A, Blázquez A, Docampo J, Ugalde C, Morán M, Arenas J, et al. Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report. Genes. 2020; 11(8):855. https://doi.org/10.3390/genes11080855
Chicago/Turabian StyleGonzález-Quintana, Adrián, Inés García-Consuegra, Amaya Belanger-Quintana, Pablo Serrano-Lorenzo, Alejandro Lucia, Alberto Blázquez, Jorge Docampo, Cristina Ugalde, María Morán, Joaquín Arenas, and et al. 2020. "Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report" Genes 11, no. 8: 855. https://doi.org/10.3390/genes11080855
APA StyleGonzález-Quintana, A., García-Consuegra, I., Belanger-Quintana, A., Serrano-Lorenzo, P., Lucia, A., Blázquez, A., Docampo, J., Ugalde, C., Morán, M., Arenas, J., & Martín, M. A. (2020). Novel NDUFA13 Mutations Associated with OXPHOS Deficiency and Leigh Syndrome: A Second Family Report. Genes, 11(8), 855. https://doi.org/10.3390/genes11080855