The Effects of Genetic and Epigenetic Alterations of BARD1 on the Development of Non-Breast and Non-Gynecological Cancers

 , , , and
, , , and
Abstract
1. Introduction
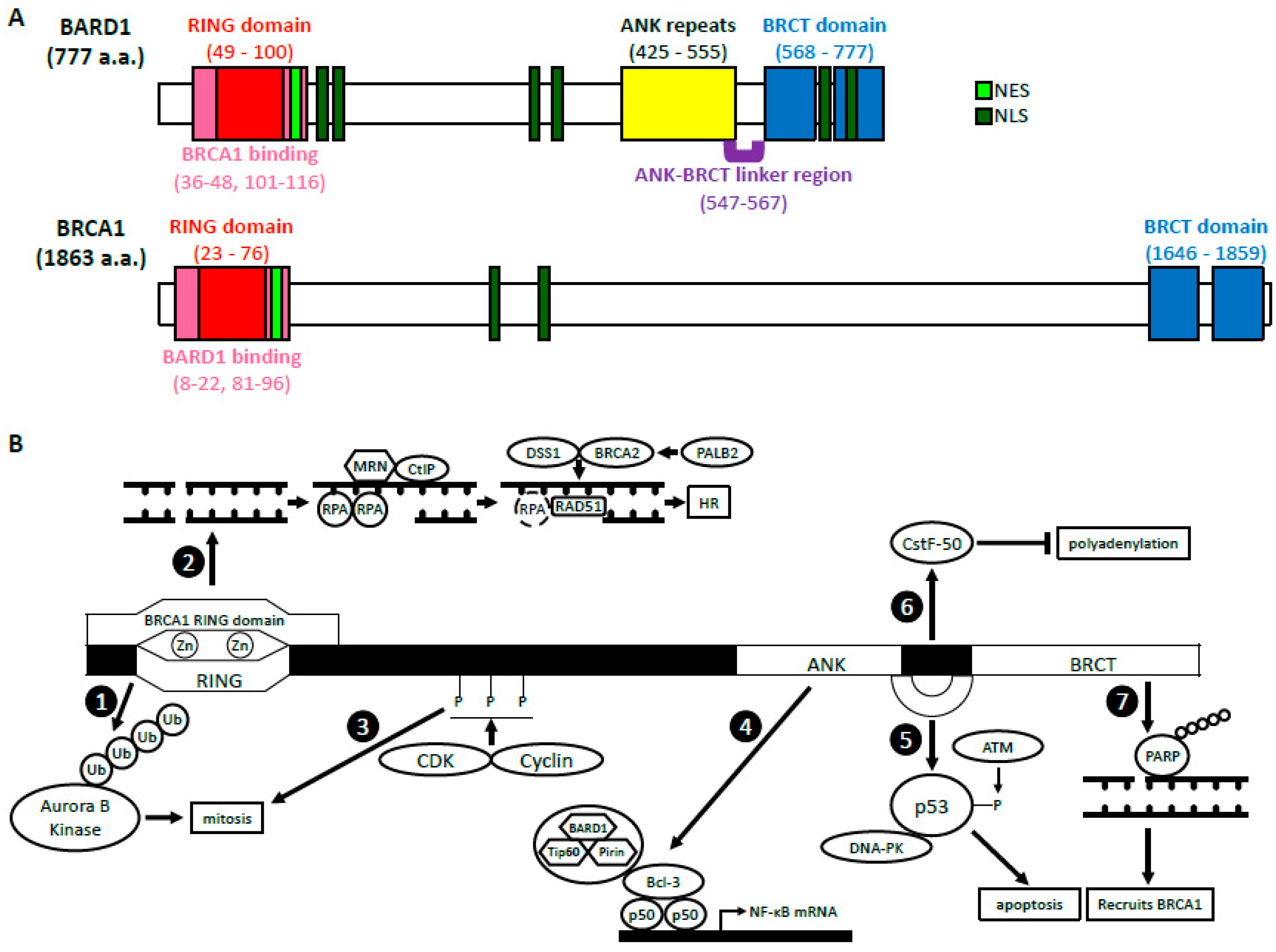
2. BRCA1-Dependent Function of BARD1
2.1. BARD1-BRCA1 E3 Ubiquitin Ligase Activity
2.2. BARD1-BRCA1 in Homologous Recombination
2.2.1. DNA End Resection
2.2.2. Presynaptic Complex Formation
2.3. BARD1-BRCA1 in Mismatch Repair
3. BRCA1-Independent Functions of BARD1
3.1. Regulation of p53 and Apoptosis
3.2. Cell Cycle Regulation/Mitosis
3.3. NF-κB
3.4. Inhibition of mRNA Processing in Response to DNA Damage
3.5. ADP Ribosylation, Poly (ADP-Ribose) Polymerase and BARD1
4. BARD1 in Non-Breast and Non-Gynecological Cancers
4.1. Neuroblastoma
4.2. Gastrointestinal Cancers
4.2.1. Normal Colon and Colorectal Cancers
4.2.2. Esophageal Squamous Cell Carcinoma
4.2.3. Hepatocellular Carcinoma
4.2.4. Pancreatic Cancer
4.3. Non-Small Cell Lung Cancer
4.4. Other Cancer Types
4.4.1. Nephroblastoma
4.4.2. Ewing Sarcoma
4.4.3. Leukemia
4.4.4. A Brief Summary of Genetic Changes in BARD1 Gene in a Variety of Cancers Identified through the Cancer Genome Atlas (TCGA) Project
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, L.C.; Wang, Z.W.; Tsan, J.T.; Spillman, M.A.; Phung, A.; Xu, X.L.; Yang, M.C.W.; Hwang, L.Y.; Bowcock, A.M.; Baer, R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat. Genet. 1996, 14, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Formicola, D.; Capasso, M. Dualistic role of BARD1 in cancer. Genes 2017, 8, 375. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Ratajska, M.; Pilyugin, M. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int. J. Biochem. Cell Biol. 2016, 72, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef]
- Birrane, G.; Varma, A.K.; Soni, A.; Ladias, J.A. Crystal structure of the BARD1 BRCT domains. Biochemistry 2007, 46, 7706–7712. [Google Scholar] [CrossRef]
- Edwards, R.A.; Lee, M.S.; Tsutakawa, S.E.; Williams, R.S.; Tainer, J.A.; Glover, J.M. The BARD1 C-terminal domain structure and interactions with polyadenylation factor CstF-50. Biochemistry 2008, 47, 11446–11456. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Schüchner, S.; Au, W.W.; Fabbro, M.; Henderson, B.R. Nuclear–cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene 2004, 23, 1809–1820. [Google Scholar] [CrossRef]
- Schüchner, S.; Tembe, V.; Rodriguez, J.A.; Henderson, B.R. Nuclear targeting and cell cycle regulatory function of human BARD1. J. Biol. Chem. 2005, 280, 8855–8861. [Google Scholar] [CrossRef] [PubMed]
- Fox, D.; Le Trong, I.; Rajagopal, P.; Brzovic, P.S.; Stenkamp, R.E.; Klevit, R.E. Crystal structure of the BARD1 ankyrin repeat domain and its functional consequences. J. Biol. Chem. 2008, 283, 21179–21186. [Google Scholar] [CrossRef] [PubMed]
- Thai, T.H.; Du, F.; Tsan, J.T.; Jin, Y.; Phung, A.; Spillman, M.A.; Massa, H.F.; Muller, C.Y.; Ashfaq, R.; Michael Mathis, J.; et al. Mutations in the BRCA1-associated RING domain (BARD1) gene in primary breast, ovarian and uterine cancers. Hum. Mol. Genet. 1998, 7, 195–202. [Google Scholar] [CrossRef]
- Karppinen, S.M.; Heikkinen, K.; Rapakko, K.; Winqvist, R. Mutation screening of the BARD1 gene: Evidence for involvement of the Cys557Ser allele in hereditary susceptibility to breast cancer. J. Med. Genet. 2004, 41, e114. [Google Scholar] [CrossRef] [PubMed]
- Ghimenti, C.; Sensi, E.; Presciuttini, S.; Brunetti, I.M.; Conte, P.; Bevilacqua, G.; Caligo, M.A. Germline mutations of the BRCA1-associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosom. Cancer 2002, 33, 235–242. [Google Scholar] [CrossRef]
- Li, L.; Ryser, S.; Dizin, E.; Pils, D.; Krainer, M.; Jefford, C.E.; Bertoni, F.; Zeillinger, R.; Irminger-Finger, I. Oncogenic BARD1 isoforms expressed in gynecological cancers. Cancer Res. 2007, 67, 11876–11885. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Pilyugin, M.; Kuester, D.; Leoni, V.P.; Li, L.; Casula, G.; Zorcolo, L.; Schneider-Stock, R.; Atzori, L.; Irminger-Finger, I. Expression of oncogenic BARD1 isoforms affects colon cancer progression and correlates with clinical outcome. Br. J. Cancer 2012, 107, 675–683. [Google Scholar] [CrossRef]
- Feki, A.; Jefford, C.E.; Berardi, P.; Wu, J.Y.; Cartier, L.; Krause, K.H.; Irminger-Finger, I. BARD1 induces apoptosis by catalysing phosphorylation of p53 by DNA-damage response kinase. Oncogene 2005, 24, 3726–3736. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, M.; Wu, W.; Nishikawa, H.; Hayami, R.; Oyake, D.; Yabuki, Y.; Fukuda, M.; Ohta, T. A truncated splice variant of human BARD1 that lacks the RING finger and ankyrin repeats. Cancer Lett. 2006, 233, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Lepore, I.; Dell’Aversana, C.; Pilyugin, M.; Conte, M.; Nebbioso, A.; De Bellis, F.; Tambaro, F.P.; Izzo, T.; Garcia-Manero, G.; Ferrara, F.; et al. HDAC inhibitors repress BARD1 isoform expression in acute myeloid leukemia cells via activation of miR-19a and/or b. PLoS ONE 2013, 8, e83018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Bianco, A.; Malkinson, A.M.; Leoni, V.P.; Frau, G.; De Rosa, N.; André, P.A.; Versace, R.; Boulvain, M.; Laurent, G.J.; et al. BARD1: An independent predictor of survival in non-small cell lung cancer. Int. J. Cancer 2012, 131, 83–94. [Google Scholar] [CrossRef]
- Ratajska, M.; Matusiak, M.; Kuzniacka, A.; Wasag, B.; Brozek, I.; Biernat, W.; Koczkowska, M.; Debniak, J.; Sniadecki, M.; Kozlowski, P.; et al. Cancer predisposing BARD1 mutations affect exon skipping and are associated with overexpression of specific BARD1 isoforms. Oncol. Rep. 2015, 34, 2609–2617. [Google Scholar] [CrossRef]
- Hashizume, R.; Fukuda, M.; Maeda, I.; Nishikawa, H.; Oyake, D.; Yabuki, Y.; Ogata, H.; Ohta, T. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by a breast cancer-derived mutation. J. Biol. Chem. 2001, 276, 14537–14540. [Google Scholar] [CrossRef]
- Xia, Y.; Pao, G.M.; Chen, H.W.; Verma, I.M.; Hunter, T. Enhancement of BRCA1 E3 ubiquitin ligase activity through direct interaction with the BARD1 protein. J. Biol. Chem. 2003, 278, 5255–5263. [Google Scholar] [CrossRef] [PubMed]
- Wu-Baer, F.; Lagrazon, K.; Yuan, W.; Baer, R. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J. Biol. Chem. 2003, 278, 34743–34746. [Google Scholar] [CrossRef]
- Nishikawa, H.; Ooka, S.; Sato, K.; Arima, K.; Okamoto, J.; Klevit, R.E.; Fukuda, M.; Ohta, T. Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J. Biol. Chem. 2004, 279, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Kleiman, F.E.; Manley, J.L.; Ouchi, T.; Pan, Z.Q. Autoubiquitination of the BRCA1*BARD1 RING ubiquitin ligase. J. Biol. Chem. 2002, 277, 22085–22092. [Google Scholar] [CrossRef]
- Hsu, L.C.; Doan, T.P.; White, R.L. Identification of a gamma-tubulin-binding domain in BRCA1. Cancer Res. 2001, 61, 7713–7718. [Google Scholar]
- Starita, L.M.; Machida, Y.; Sankaran, S.; Elias, J.E.; Griffin, K.; Schlegel, B.P.; Gygi, S.P.; Parvin, J.D. BRCA1-dependent ubiquitination of gamma-tubulin regulates centrosome number. Mol. Cell Biol. 2004, 24, 8457–8466. [Google Scholar] [CrossRef]
- Sankaran, S.; Starita, L.M.; Groen, A.C.; Ko, M.J.; Parvin, J.D. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol. Cell Biol. 2005, 25, 8656–8668. [Google Scholar] [CrossRef]
- Sankaran, S.; Crone, D.E.; Palazzo, R.E.; Parvin, J.D. BRCA1 regulates gamma-tubulin binding to centrosomes. Cancer Biol. Ther. 2007, 6, 1853–1857. [Google Scholar] [CrossRef][Green Version]
- Zou, J.; Zhang, D.; Qin, G.; Chen, X.; Wang, H.; Zhang, D. BRCA1 and FancJ cooperatively promote interstrand crosslinker induced centrosome amplification through the activation of polo-like kinase 1. Cell Cycle 2014, 13, 3685–3697. [Google Scholar] [CrossRef]
- Joukov, V.; Groen, A.C.; Prokhorova, T.; Gerson, R.; White, E.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006, 127, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Ryser, S.; Dizin, E.; Jefford, C.E.; Delaval, B.; Gagos, S.; Christodoulidou, A.; Krause, K.H.; Birnbaum, D.; Irminger-Finger, I. Distinct roles of BARD1 isoforms in mitosis: Full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 2009, 69, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Delaval, B.; Ferrand, A.; Conte, N.; Larroque, C.; Hernandez-Verdun, D.; Prigent, C.; Birnbaum, D. Aurora B-TACC1 protein complex in cytokinesis. Oncogene 2004, 23, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Thakar, A.; Parvin, J.; Zlatanova, J. BRCA1/BARD1 E3 ubiquitin ligase can modify histones H2A and H2B in the nucleosome particle. J. Biomol. Struct. Dyn. 2010, 27, 399–406. [Google Scholar] [CrossRef]
- Zhu, Q.; Pao, G.M.; Huynh, A.M.; Suh, H.; Tonnu, N.; Nederlof, P.M.; Gage, F.H.; Verma, I.M. BRCA1 tumour suppression occurs via heterochromatin-mediated silencing. Nature 2011, 477, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Densham, R.M.; Garvin, A.J.; Stone, H.R.; Strachan, J.; Baldock, R.A.; Daza-Martin, M.; Fletcher, A.; Blair-Reid, S.; Beesley, J.; Johal, B.; et al. Human BRCA1-BARD1 ubiquitin ligase activity counteracts chromatin barriers to DNA resection. Nat. Struct. Mol. Biol. 2016, 23, 647–655. [Google Scholar] [CrossRef]
- Stewart, M.D.; Zelin, E.; Dhall, A.; Walsh, T.; Upadhyay, E.; Corn, J.E.; Chatterjee, C.; King, M.C.; Klevit, R.E. BARD1 is necessary for ubiquitylation of nucleosomal histone H2A and for transcriptional regulation of estrogen metabolism genes. Proc. Natl. Acad. Sci. USA 2018, 115, 1316–1321. [Google Scholar] [CrossRef]
- Calvo, V.; Beato, M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011, 71, 3422–3431. [Google Scholar] [CrossRef]
- Noordermeer, S.M. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Anand, R.; Ranjha, L.; Cannavo, E.; Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 2016, 64, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, A.; Lopez-Saavedra, A.; Huertas, P. BRCA1 accelerates CtIP-mediated DNA-end resection. Cell Rep. 2014, 9, 451–459. [Google Scholar] [CrossRef]
- Reczek, C.R.; Szabolcs, M.; Stark, J.M.; Ludwig, T.; Baer, R. The interaction between CtIP and BRCA1 is not essential for resection-mediated DNA repair or tumor suppression. J. Cell Biol. 2013, 201, 693–707. [Google Scholar] [CrossRef]
- Polato, F.; Callen, E.; Wong, N.; Faryabi, R.; Bunting, S.; Chen, H.T.; Kozak, M.; Kruhlak, M.J.; Reczek, C.R.; Lee, W.H.; et al. CtIP-mediated resection is essential for viability and can operate independently of BRCA1. J. Exp. Med. 2014, 211, 1027–1036. [Google Scholar] [CrossRef]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef]
- Bhat, K.P.; Cortez, D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018, 25, 446–453. [Google Scholar] [CrossRef]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Ma, C.J.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]
- Sy, S.M.; Huen, M.S.; Chen, J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. USA 2009, 106, 7155–7160. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 9, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous recombination and human health: The roles of BRCA1, BRCA2, and associated proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, H.; Guerrette, S.; Chen, J.; Mazurek, A.; Wilson, T.; Slupianek, A.; Skorski, T.; Fishel, R.; Greene, M.I. Adenosine nucleotide modulates the physical interaction between hMSH2 and BRCA1. Oncogene 2001, 20, 4640–4649. [Google Scholar] [CrossRef][Green Version]
- Romeo, F.; Falbo, L.; Di Sanzo, M.; Misaggi, R.; Faniello, M.C.; Viglietto, G.; Cuda, G.; Costanzo, F.; Quaresima, B. BRCA1 is required for hMLH1 stabilization following doxorubicin-induced DNA damage. Int. J. Biochem. Cell Biol. 2011, 43, 1754–1763. [Google Scholar] [CrossRef]
- Maresca, L.; Spugnesi, L.; Lodovichi, S.; Cozzani, C.; Naccarato, A.G.; Tancredi, M.; Collavoli, A.; Falaschi, E.; Rossetti, E.; Aretini, P.; et al. MSH2 role in BRCA1-driven tumorigenesis: A preliminary study in yeast and in human tumors from BRCA1-VUS carriers. Eur. J. Med. Genet. 2015, 58, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Phelan, C.M.; Iqbal, J.; Lynch, H.T.; Lubinski, J.; Gronwald, J.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; et al. Incidence of colorectal cancer in BRCA1 and BRCA2 mutation carriers: Results from a follow-up study. Br. J. Cancer 2014, 110, 530–534. [Google Scholar] [CrossRef]
- Irminger-Finger, I.; Leung, W.C.; Li, J.; Dubois-Dauphin, M.; Harb, J.; Feki, A.; Jefford, C.E.; Soriano, J.V.; Jaconi, M.; Montesano, R.; et al. Identification of BARD1 as mediator between proapoptotic stress and p53-dependent apoptosis. Mol. Cell 2001, 8, 1255–1266. [Google Scholar] [CrossRef]
- Jefford, C.E.; Feki, A.; Harb, J.; Krause, K.H.; Irminger-Finger, I. Nuclear–cytoplasmic translocation of BARD1 is linked to its apoptotic activity. Oncogene 2004, 23, 3509–3520. [Google Scholar] [CrossRef]
- Tembe, V.; Martino-Echarri, E.; Marzec, K.A.; Mok, M.T.; Brodie, K.M.; Mills, K.; Lei, Y.; DeFazio, A.; Rizos, H.; Kettle, E.; et al. The BARD1 BRCT domain contributes to p53 binding, cytoplasmic and mitochondrial localization, and apoptotic function. Cell. Signal. 2015, 27, 1763–1771. [Google Scholar] [CrossRef]
- Yim, E.-K.; Lee, K.-H.; Myeong, J.; Tong, S.-Y.; Um, S.J.; Park, J.-S. Novel interaction between HPV E6 and BARD1 (BRCA1-associated ring domain 1) and its biologic roles. DNA Cell Biol. 2007, 26, 753–761. [Google Scholar] [CrossRef]
- Tembe, V.; Henderson, B.R. BARD1 translocation to mitochondria correlates with Bax oligomerization, loss of mitochondrial membrane potential, and apoptosis. J. Biol. Chem. 2007, 282, 20513–20522. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yang, E.S.; Jiang, G.; Nowsheen, S.; Wang, H.; Wang, T.; Wang, Y.; Billheimer, D.; Chakravarthy, A.B.; Brown, M.; et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Res. 2011, 71, 5546–5557. [Google Scholar] [CrossRef] [PubMed]
- Fabbro, M.; Savage, K.; Hobson, K.; Deans, A.J.; Powell, S.N.; McArthur, G.A.; Khanna, K.K. BRCA1-BARD1 complexes are required for p53Ser-15 phosphorylation and a G1/S arrest following ionizing radiation-induced DNA damage. J. Biol. Chem. 2004, 279, 31251–31258. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D.; Xu, H.; Baer, R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 2004, 279, 33909–33918. [Google Scholar] [CrossRef] [PubMed]
- Irminger-Finger, I.; Soriano, J.V.; Vaudan, G.; Montesano, R.; Sappino, A.P. In vitro repression of Brca1-associated RING domain gene, Bard1, induces phenotypic changes in mammary epithelial cells. J. Cell Biol. 1998, 143, 1329–1339. [Google Scholar] [CrossRef]
- Hayami, R.; Sato, K.; Wu, W.; Nishikawa, T.; Hiroi, J.; Ohtani-Kaneko, R.; Fukuda, M.; Ohta, T. Down-regulation of BRCA1-BARD1 ubiquitin ligase by CDK2. Cancer Res. 2005, 65, 6–10. [Google Scholar]
- Dechend, R.; Hirano, F.; Lehmann, K.; Heissmeyer, V.; Ansieau, S.; Wulczyn, F.G.; Scheidereit, C.; Leutz, A. The Bcl-3 oncoprotein acts as a bridging factor between NF-κB/Rel and nuclear co-regulators. Oncogene 1999, 18, 3316–3323. [Google Scholar] [CrossRef]
- Irminger-Finger, I.; Leung, W.-C. BRCA1-dependent and independent functions of BARD1. Int. J. Biochem. Cell Biol. 2002, 34, 582–587. [Google Scholar] [CrossRef]
- Benezra, M.; Chevallier, N.; Morrison, D.J.; MacLachlan, T.K.; El-Deiry, W.S.; Licht, J.D. BRCA1 augments transcription by the NF-κB transcription factor by binding to the Rel domain of the p65/RelA subunit. J. Biol. Chem. 2003, 278, 26333–26341. [Google Scholar] [CrossRef]
- Kleiman, F.E.; Manley, J.L. Functional interaction of BRCA1-associated BARD1 with polyadenylation factor CstF-50. Science 1999, 285, 1576–1579. [Google Scholar] [CrossRef]
- Cevher, M.A.; Kleiman, F.E. Connections between 3’-end processing and DNA damage response. Wiley Interdiscip. Rev. RNA 2010, 1, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, F.E.; Manley, J.L. The BARD1-CstF-50 interaction links mRNA 3’ end formation to DNA damage and tumor suppression. Cell 2001, 104, 743–753. [Google Scholar] [CrossRef]
- Kleiman, F.E.; Wu-Baer, F.; Fonseca, D.; Kaneko, S.; Baer, R.; Manley, J.L. BRCA1/BARD1 inhibition of mRNA 3’ processing involves targeted degradation of RNA polymerase II. Genes Dev. 2005, 19, 1227–1237. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Li, H.; Cevher, M.; Parmelee, A.; Fonseca, D.; Kleiman, F.E.; Lee, S.B. DNA Damage–Induced BARD1 Phosphorylation Is Critical for the Inhibition of Messenger RNA Processing by BRCA1/BARD1 Complex. Cancer Res. 2006, 66, 4561–4565. [Google Scholar] [CrossRef] [PubMed]
- Nazeer, F.I.; Devany, E.; Mohammed, S.; Fonseca, D.; Akukwe, B.; Taveras, C.; Kleiman, F.E. p53 inhibits mRNA 3’ processing through its interaction with the CstF/BARD1 complex. Oncogene 2011, 30, 3073–3083. [Google Scholar] [CrossRef][Green Version]
- Hottiger, M.O. Poly(ADP-ribose) polymerase inhibitor therapeutic effect: Are we just scratching the surface? Expert Opin. Ther. Targets 2015, 19, 1149–1152. [Google Scholar] [CrossRef][Green Version]
- Azarm, K.; Smith, S. Nuclear PARPs and genome integrity. Genes Dev. 2020, 34, 285–301. [Google Scholar] [CrossRef]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef]
- Bürkle, A. Physiology and pathophysiology of poly (ADP-ribosyl) ation. Bioessays 2001, 23, 795–806. [Google Scholar] [CrossRef]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly (ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly (ADP-ribose) glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar] [CrossRef]
- Mateo, J.; Lord, C.J.; Serra, V.; Tutt, A.; Balmaña, J.; Castroviejo-Bermejo, M.; Cruz, C.; Oaknin, A.; Kaye, S.B.; De Bono, J.S. A decade of clinical development of PARP inhibitors in perspective. Ann. Oncol. 2019, 30, 1437–1447. [Google Scholar] [CrossRef]
- Colon, N.C.; Chung, D.H. Neuroblastoma. Adv. Pediatr. 2011, 58, 297–311. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Devoto, M.; Hou, C.; Asgharzadeh, S.; Glessner, J.T.; Attiyeh, E.F.; Mosse, Y.P.; Kim, C.; Diskin, S.J.; Cole, K.A.; et al. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 2009, 41, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Capasso, M.; Diskin, S.J.; Totaro, F.; Longo, L.; Mariano, M.D.; Russo, R.; Cimmino, F.; Hakonarson, H.; Tonini, G.P.; Devoto, M.; et al. Replication of GWAS-identified neuroblastoma risk loci strengthens the role of BARD1 and affirms the cumulative effect of genetic variations on disease susceptibility. Carcinogenesis 2013, 34, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Latorre, V.; Diskin, S.J.; Diamond, M.A.; Zhang, H.; Hakonarson, H.; Maris, J.M.; Devoto, M. Replication of neuroblastoma SNP association at the BARD1 locus in African-Americans. Cancer Epidemiol. Biomark. Prev. 2012, 21, 658–663. [Google Scholar] [CrossRef]
- Shi, J.; Yu, Y.; Jin, Y.; Lu, J.; Zhang, J.; Wang, H.; Han, W.; Chu, P.; Tai, J.; Chen, F.; et al. Functional Polymorphisms in BARD1 association with neuroblastoma in a regional Han Chinese population. J. Cancer 2019, 10, 2153–2160. [Google Scholar] [CrossRef]
- Zhang, R.; Zou, Y.; Zhu, J.; Zeng, X.; Yang, T.; Wang, F.; He, J.; Xia, H. The Association between GWAS-identified BARD1 Gene SNPs and Neuroblastoma Susceptibility in a Southern Chinese Population. Int. J. Med. Sci. 2016, 13, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen, L.B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef]
- Sakka, L.; Delétage, N.; Chalus, M.; Aissouni, Y.; Sylvain-Vidal, V.; Gobron, S.; Coll, G. Assessment of citalopram and escitalopram on neuroblastoma cell lines. Cell toxicity and gene modulation. Oncotarget 2017, 8, 42789–42807. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Avitabile, M.; Diskin, S.J.; Vaksman, Z.; Pignataro, P.; Formicola, D.; Cardinale, A.; Testori, A.; Koster, J.; de Torres, C.; et al. Fine mapping of 2q35 high-risk neuroblastoma locus reveals independent functional risk variants and suggests full-length BARD1 as tumor-suppressor. Int. J. Cancer 2018, 143, 2828–2837. [Google Scholar] [CrossRef] [PubMed]
- Tonini, G.P.; Capasso, M. Genetic predisposition and chromosome instability in neuroblastoma. Cancer Metastasis Rev. 2020, 39, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, D.A.; Truong, B.; Russ, D.; DuBois, S.G.; Vaksman, Z.; Mosse, Y.P.; Diskin, S.J.; Maris, J.M.; Matthay, K.K. Differences in genomic profiles and outcomes between thoracic and adrenal neuroblastoma. J. Natl. Cancer Inst. 2019, 111, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Genome-wide pathway analysis in neuroblastoma. Tumour Biol. 2014, 35, 3471–3485. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, F.; Avitabile, M.; Lasorsa, V.A.; Pezone, L.; Cardinale, A.; Montella, A.; Cantalupo, S.; Iolascon, A.; Capasso, M. Functional characterization of full-length BARD1 strengthens its role as a tumor suppressor in neuroblastoma. J. Cancer 2020, 11, 1495–1504. [Google Scholar] [CrossRef]
- Takagi, M.; Yoshida, M.; Nemoto, Y.; Tamaichi, H.; Tsuchida, R.; Seki, M.; Uryu, K.; Nishii, R.; Miyamoto, S.; Saito, M.; et al. Loss of DNA Damage Response in Neuroblastoma and Utility of a PARP Inhibitor. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Hisamuddin, I.M.; Yang, V.W. Genetics of colorectal cancer. Med. Gen. Med. 2004, 6, 13. [Google Scholar]
- Sporn, J.C.; Hothorn, T.; Jung, B. BARD1 expression predicts outcome in colon cancer. Clin. Cancer Res. 2011, 17, 5451–5462. [Google Scholar] [CrossRef] [PubMed]
- Ozden, O.; Bishehsari, F.; Bauer, J.; Park, S.H.; Jana, A.; Baik, S.H.; Sporn, J.C.; Staudacher, J.J.; Yazici, C.; Krett, N.; et al. Expression of an oncogenic BARD1 splice variant impairs homologous recombination and predicts response to PARP-1 inhibitor therapy in colon cancer. Sci. Rep. 2016, 6, 26273. [Google Scholar] [CrossRef] [PubMed]
- Pilyugin, M.; Irminger-Finger, I. Long non-coding RNA and microRNAs might act in regulating the expression of BARD1 mRNAs. Int. J. Biochem. Cell Biol. 2014, 54, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Gautier, F.; Irminger-Finger, I.; Grégoire, M.; Meflah, K.; Harb, J. Identification of an apoptotic cleavage product of BARD1 as an autoantigen: A potential factor in the antitumoral response mediated by apoptotic bodies. Cancer Res. 2000, 60, 6895–6900. [Google Scholar]
- Esteban-Jurado, C.; Vila-Casadesús, M.; Garre, P.; Lozano, J.J.; Pristoupilova, A.; Beltran, S.; Muñoz, J.; Ocaña, T.; Balaguer, F.; López-Cerón, M.; et al. Whole-exome sequencing identifies rare pathogenic variants in new predisposition genes for familial colorectal cancer. Genet. Med. 2015, 17, 131142. [Google Scholar] [CrossRef]
- Toh, M.R.; Chong, S.T.; Chan, S.H.; Low, C.E.; Ishak, N.D.B.; Lim, J.Q.; Courtney, E.; Ngeow, J. Functional analysis of clinical BARD1 germline variants. Cold Spring Harb. Mol. Case Stud. 2019, 5. [Google Scholar] [CrossRef]
- Huang, F.L.; Yu, S.J. Esophageal cancer: Risk factors, genetic association, and treatment. Asian J. Surg. 2018, 41, 210–215. [Google Scholar] [CrossRef]
- Yang, Q.; Pan, Q.; Li, C.; Xu, Y.; Wen, C.; Sun, F. NRAGE is involved in homologous recombination repair to resist the DNA-damaging chemotherapy and composes a ternary complex with RNF8-BARD1 to promote cell survival in squamous esophageal tumorigenesis. Cell Death Differ. 2016, 23, 1406–1416. [Google Scholar] [CrossRef]
- Hou, S.; Jin, W.; Xiao, W.; Deng, B.; Wu, D.; Zhi, J.; Wu, K.; Cao, X.; Chen, S.; Ding, Y.; et al. Integrin α5 promotes migration and cisplatin resistance in esophageal squamous cell carcinoma cells. Am. J. Cancer Res. 2019, 9, 2774–2788. [Google Scholar]
- Lubecka, K.; Flower, K.; Beetch, M.; Qiu, J.; Kurzava, L.; Buvala, H.; Ruhayel, A.; Gawrieh, S.; Liangpunsakul, S.; Gonzalez, T.; et al. Loci-specific differences in blood DNA methylation in HBV-negative populations at risk for hepatocellular carcinoma development. Epigenetics 2018, 13, 605–626. [Google Scholar] [CrossRef]
- Liao, Y.; Yuan, S.; Chen, X.; Zhu, P.; Li, J.; Qin, L.; Liao, W. Up-regulation of BRCA1-associated RING Domain 1 promotes hepatocellular carcinoma progression by targeting Akt signaling. Sci. Rep. 2017, 7, 7649. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.C.; Selander, I.; Connor, A.A.; Selvarajah, S.; Borgida, A.; Briollais, L.; Petersen, G.M.; Lerner-Ellis, J.; Holter, S.; Gallinger, S. Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 2015, 148, 556–564. [Google Scholar] [CrossRef]
- Chaffee, K.G.; Oberg, A.L.; McWilliams, R.R.; Majithia, N.; Allen, B.A.; Kidd, J.; Singh, N.; Hartman, A.R.; Wenstrup, R.J.; Petersen, G.M. Prevalence of germ-line mutations in cancer genes among pancreatic cancer patients with a positive family history. Genet. Med. 2018, 20, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Pugh, T.J.; Morozova, O.; Attiyeh, E.F.; Asgharzadeh, S.; Wei, J.S.; Auclair, D.; Carter, S.L.; Cibulskis, K.; Hanna, M.; Kiezun, A.; et al. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 2013, 45, 279–284. [Google Scholar] [CrossRef]
- Lasorsa, V.A.; Formicola, D.; Pignataro, P.; Cimmino, F.; Calabrese, F.M.; Mora, J.; Esposito, M.R.; Pantile, M.; Zanon, C.; De Mariano, M.; et al. Exome and deep sequencing of clinically aggressive neuroblastoma reveal somatic mutations that affect key pathways involved in cancer progression. Oncotarget 2016, 7, 21840–21852. [Google Scholar] [CrossRef]
- Tan, A.C.; Fan, J.B.; Karikari, C.; Bibikova, M.; Wickham Garcia, E.; Zhou, L.; Barker, D.; Serre, D.; Feldmann, G.; Hruban, R.H.; et al. Allele-specific expression in the germline of patients with familial pancreatic cancer: An unbiased approach to cancer gene discovery. Cancer Biol. Ther. 2008, 7, 135–144. [Google Scholar] [CrossRef]
- Klöppel, G. Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr. Relat. Cancer 2011, 18 (Suppl. S1), S1–S16. [Google Scholar] [CrossRef]
- Szybowska, M.; Mete, O.; Weber, E.; Silver, J.; Kim, R.H. Neuroendocrine neoplasms associated with germline pathogenic variants in the homologous recombination pathway. Endocr. Pathol. 2019, 30, 237–245. [Google Scholar] [CrossRef]
- Zhang, T.; Wang, X.; Yue, Z. Identification of candidate genes related to pancreatic cancer based on analysis of gene co-expression and protein-protein interaction network. Oncotarget 2017, 8, 71105–71116. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Vlastos, A.T.; Pelte, M.F.; Caligo, M.A.; Bianco, A.; Krause, K.H.; Laurent, G.J.; Irminger-Finger, I. Aberrant expression of BARD1 in breast and ovarian cancers with poor prognosis. Int. J. Cancer 2006, 118, 1215–1226. [Google Scholar] [CrossRef]
- Pierre-Alain, A.; Prele, C.M.; Vierkotten, S.; Carnesecchi, S.; Donati, Y.; Chambers, R.C.; Pache, J.C.; Crestani, B.; Barazzone-Argiroffo, C.; Konigshoff, M.; et al. BARD1 mediates TGF-β signaling in pulmonary fibrosis. Respir. Res. 2015, 16, 118. [Google Scholar]
- Pilyugin, M.; Descloux, P.; André, P.A.; Laszlo, V.; Dome, B.; Hegedus, B.; Sardy, S.; Janes, S.; Bianco, A.; Laurent, G.J.; et al. BARD1 serum autoantibodies for the detection of lung cancer. PLoS ONE 2017, 12, e0182356. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.P.; Hu, Y.C.; Xie, Y.; Jin, F.; Song, Z.Q.; Liu, X.D.; Ma, T.; Zhou, P.K. MLN4924 suppresses the BRCA1 complex and synergizes with PARP inhibition in NSCLC cells. Biochem. Biophys. Res. Commun. 2017, 483, 223–229. [Google Scholar] [CrossRef]
- Ko, E.Y.; Ritchey, M.L. Current management of Wilms’ tumor in children. J. Pediatr. Urol. 2009, 5, 56–65. [Google Scholar] [CrossRef]
- Fu, W.; Zhu, J.; Xiong, S.W.; Jia, W.; Zhao, Z.; Zhu, S.B.; Hu, J.H.; Wang, F.H.; Xia, H.; He, J.; et al. BARD1 Gene Polymorphisms Confer Nephroblastoma Susceptibility. EBioMedicine 2017, 16, 101–105. [Google Scholar] [CrossRef]
- Choi, E.Y.; Gardner, J.M.; Lucas, D.R.; McHugh, J.B.; Patel, R.M. Ewing sarcoma. Semin. Diagn. Pathol. 2014, 31, 39–47. [Google Scholar] [CrossRef]
- Spahn, L.; Petermann, R.; Siligan, C.; Schmid, J.A.; Aryee, D.N.; Kovar, H. Interaction of the EWS NH2 terminus with BARD1 links the Ewing’s sarcoma gene to a common tumor suppressor pathway. Cancer Res. 2002, 62, 4583–4587. [Google Scholar]
- Venier, R.E.; Maurer, L.M.; Kessler, E.M.; Ranganathan, S.; McGough, R.L.; Weiss, K.R.; Malek, M.M.; Meade, J.; Tersak, J.M.; Bailey, K.M. A germline BARD1 mutation in a patient with Ewing sarcoma: Implications for familial testing and counseling. Pediatr. Blood Cancer 2019, 66, e27824. [Google Scholar] [CrossRef]
- Griffin, J.D.; Löwenberg, B. Clonogenic cells in acute myeloblastic leukemia. Blood 1986, 68, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Adamovich, A.I.; Banerjee, T.; Wingo, M.; Duncan, K.; Ning, J.; Rodrigues, F.M.; Huang, K.L.; Lee, C.; Chen, F.; Ding, L.; et al. Functional analysis of BARD1 missense variants in homology-directed repair and damage sensitivity. PLoS Genet. 2019, 15, e1008049. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Marquart, L.; McGuffog, L.; Healey, S.; Sinilnikova, O.; Wan, F.; Chen, X.; Beesley, J.; Singer, C.F.; Dressler, A.C.; et al. Common genetic variation at BARD1 is not associated with breast cancer risk in BRCA1 or BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2011, 20, 1032–1038. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Domchek, S.M.; Wan, F.; Chuai, S.; Friebel, T.M.; Panossian, S.; Spurdle, A.; Chenevix-Trench, G.; Singer, C.F.; et al. Modification of ovarian cancer risk by BRCA1/2-interacting genes in a multicenter cohort of BRCA1/2 mutation carriers. Cancer Res. 2009, 69, 5801–5810. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, Y.; Boujemaa, M.; Rekaya, M.B.; Hamda, C.B.; Mighri, N.; El Benna, H.; Mejri, N.; Labidi, S.; Daoud, N.; Naouali, C.; et al. Family specific genetic predisposition to breast cancer: Results from Tunisian whole exome sequenced breast cancer cases. J. Transl. Med. 2018, 16, 158. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Sun, X.; He, Y.; Li, J.; Guo, X. A cross-sectional study of associations between nonsynonymous mutations of the BARD1 gene and breast cancer in Han Chinese women. Asia Pac. J. Public Health 2013, 25 (Suppl. S4), 8S–14S. [Google Scholar] [CrossRef]
- Huo, X.; Hu, Z.; Zhai, X.; Wang, Y.; Wang, S.; Wang, X.; Qin, J.; Chen, W.; Jin, G.; Liu, J.; et al. Common non-synonymous polymorphisms in the BRCA1 Associated RING Domain (BARD1) gene are associated with breast cancer susceptibility: A case-control analysis. Breast Cancer Res. Treat. 2007, 102, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Vahteristo, P.; Syrjäkoski, K.; Heikkinen, T.; Eerola, H.; Aittomäki, K.; von Smitten, K.; Holli, K.; Blomqvist, C.; Kallioniemi, O.P.; Nevanlinna, H. BARD1 variants Cys557Ser and Val507Met in breast cancer predisposition. Eur. J. Hum. Genet. 2006, 14, 167–172. [Google Scholar] [CrossRef]
- Ishitobi, M.; Miyoshi, Y.; Hasegawa, S.; Egawa, C.; Tamaki, Y.; Monden, M.; Noguchi, S. Mutational analysis of BARD1 in familial breast cancer patients in Japan. Cancer Lett. 2003, 200, 1–7. [Google Scholar] [CrossRef]
- Zhou, X.; Han, S.; Wang, S.; Chen, X.; Dong, J.; Shi, X.; Xia, Y.; Wang, X.; Hu, Z.; Shen, H. Polymorphisms in HPV E6/E7 protein interacted genes and risk of cervical cancer in Chinese women: A case-control analysis. Gynecol. Oncol. 2009, 114, 327–331. [Google Scholar] [CrossRef] [PubMed]
- De Brakeleer, S.; De Greve, J.; Desmedt, C.; Joris, S.; Sotiriou, C.; Piccart, M.; Pauwels, I.; Teugels, E. Frequent incidence of BARD1-truncating mutations in germline DNA from triple-negative breast cancer patients. Clin. Genet. 2016, 89, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.; Tatro, M.; Blackburn, P.; Hines, S.; Atwal, P.S. Nonsense variant c.1921C>T in a patient with recurrent breast cancer. Clin. Case Rep. 2017, 5, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Feliubadaló, L.; Tonda, R.; Gausachs, M.; Trotta, J.R.; Castellanos, E.; López-Doriga, A.; Teulé, À.; Tornero, E.; Del Valle, J.; Gel, B.; et al. Benchmarking of Whole Exome Sequencing and Ad Hoc Designed Panels for Genetic Testing of Hereditary Cancer. Sci. Rep. 2017, 7, 37984. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [PubMed]
- González-Rivera, M.; Lobo, M.; López-Tarruella, S.; Jerez, Y.; del Monte-Millán, M.; Massarrah, T.; Ramos-Medina, R.; Ocaña, I.; Picornell, A.; Garzón, S.S.; et al. Frequency of germline DNA genetic findings in an unselected prospective cohort of triple-negative breast cancer patients participating in a platinum-based neoadjuvant chemotherapy trial. Breast Cancer Res. Treat. 2016, 156, 507–515. [Google Scholar] [CrossRef]
- Suszynska, M.; Kluzniak, W.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Gronwald, J.; Debniak, T.; Szwiec, M.; Ratajska, M.; Klonowska, K.; et al. Is A Low/Moderate Breast Cancer Risk Gene: Evidence Based on An. Association Study of the Central European p.Q564X Recurrent Mutation. Cancers 2019, 11, 740. [Google Scholar]
- Klonowska, K.; Ratajska, M.; Czubak, K.; Kuzniacka, A.; Brozek, I.; Koczkowska, M.; Sniadecki, M.; Debniak, J.; Wydra, D.; Balut, M.; et al. Analysis of large mutations in BARD1 in patients with breast and/or ovarian cancer: The Polish population as an example. Sci. Rep. 2015, 5, 10424. [Google Scholar] [CrossRef]
- De Brakeleer, S.; De Grève, J.; Loris, R.; Janin, N.; Lissens, W.; Sermijn, E.; Teugels, E. Cancer predisposing missense and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast cancer families. Hum. Mutat. 2010, 31, E1175–E1185. [Google Scholar] [CrossRef]
- Sauer, M.K.; Andrulis, I.L. Identification and characterization of missense alterations in the BRCA1 associated RING domain (BARD1) gene in breast and ovarian cancer. J. Med. Genet. 2005, 42, 633–638. [Google Scholar] [CrossRef]
- Cury, N.M.; Brotto, D.B.; de Araujo, L.F.; Rosa, R.C.A.; Texeira, L.A.; Plaça, J.R.; Marques, A.A.; Peronni, K.C.; de Cássia Ruy, P.; Molfetta, G.A.; et al. Germline variants in DNA repair genes associated with hereditary breast and ovarian cancer syndrome: Analysis of a 21 gene panel in the Brazilian population. BMC Med. Genom. 2020, 13, 21. [Google Scholar]



{kind=link}
{kind=link}
| Neuroblastoma | ||||||||
|---|---|---|---|---|---|---|---|---|
| SNP | Susceptibility to NB | Location | Allelic Change | Function | Mechanism | Associates with | Population | Linkage Disequilibrium |
| rs6435862 | ↑ | Intron 1 | T > G | Removes exons 2 and 3 and produces the oncogenic BARD1β isoform [90] | BARD1β interacts with and stabilizes Aurora kinase A and B (independent of p53 and BRCA1) [90] | UK, AA/US: high-risk [85,87] EA/US: high-risk [85,86], stage 4N, MYCN amp, age > 18 mos. [86] Chinese: adrenal origin, stage IV, stage III & IV, age > 12 mos. [88,89] | European Americans [85,86], UK Caucasians [85], Italians [86], African Americans [87], Han Chinese [88,89] | |
| rs3768716 | ↑ | Intronic | A > G | EA/US, UK, Italians: high-risk [85,86] Chinese: adrenal origin, stage III & IV [88,89], age > 12 mos. [89] | European Americans [85,86], UK Caucasians [85], Italians [86], African Americans [87], Han Chinese [88,89] | |||
| rs17489363 | ↑ | 5′ UTR (promoter region) | C > T [92] G > A [88] | Decreases mRNA expression of BARD1-FL [88,92] | Through the binding of HSF1 [92] | EA/US, AA/US, Italians: high-risk [92] Chinese: adrenal origin [88] | European Americans, African Americans, Italians, Spaniards [92], Han Chinese [88] | rs6720708 [94] |
| rs6720708 | ↑ | C > T | EA/US: adrenal origin [94] | European Americans [94] | rs17489363 [94] | |||
| rs7585356 | ↓ | 3′ UTR | G > A | Increases mRNA expression of BARD1-FL [86] | EA/US: high-risk, stage 4N, MYCN amp, age < 18 mos. [86] Italians: stage 4N, age < 18 mos. [86] Chinese: females [89] | European Americans [85,86], Italians [86], Han Chinese [89] | rs16852600 (intronic) [85,95] | |
| rs1048108 | ↓ | Exon 1 (P24S) | C>T [85,86] G>A [92] | Negatively regulates cellular development and modulates development and apoptosis [95] | EA/US, Italians, AA/US: high-risk [92] | European Americans [85,92], Italians [86,92], African Americans, Spaniards [92] | ||
| rs16852804 | ↑ | Intronic | C > T | African Americans [87] | ||||
| rs7599060 | ↑ | Intronic | G > A | AA/US: high-risk [87] | African Americans [87] | |||
| rs373888 | ↑ | Exon 10 (R658C) | C > T | Chinese: adrenal origin [88] | Han Chinese [88] | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watters, A.K.; Seltzer, E.S.; MacKenzie, D., Jr.; Young, M.; Muratori, J.; Hussein, R.; Sodoma, A.M.; To, J.; Singh, M.; Zhang, D. The Effects of Genetic and Epigenetic Alterations of BARD1 on the Development of Non-Breast and Non-Gynecological Cancers. Genes 2020, 11, 829. https://doi.org/10.3390/genes11070829
Watters AK, Seltzer ES, MacKenzie D Jr., Young M, Muratori J, Hussein R, Sodoma AM, To J, Singh M, Zhang D. The Effects of Genetic and Epigenetic Alterations of BARD1 on the Development of Non-Breast and Non-Gynecological Cancers. Genes. 2020; 11(7):829. https://doi.org/10.3390/genes11070829
Chicago/Turabian StyleWatters, Andrea K., Emily S. Seltzer, Danny MacKenzie, Jr., Melody Young, Jonathan Muratori, Rama Hussein, Andrej M. Sodoma, Julie To, Manrose Singh, and Dong Zhang. 2020. "The Effects of Genetic and Epigenetic Alterations of BARD1 on the Development of Non-Breast and Non-Gynecological Cancers" Genes 11, no. 7: 829. https://doi.org/10.3390/genes11070829
APA StyleWatters, A. K., Seltzer, E. S., MacKenzie, D., Jr., Young, M., Muratori, J., Hussein, R., Sodoma, A. M., To, J., Singh, M., & Zhang, D. (2020). The Effects of Genetic and Epigenetic Alterations of BARD1 on the Development of Non-Breast and Non-Gynecological Cancers. Genes, 11(7), 829. https://doi.org/10.3390/genes11070829