Evidence for Dosage Compensation in Coccinia grandis, a Plant with a Highly Heteromorphic XY System

 , , ,
, , ,
Abstract
1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. RNA Sequencing
2.3. De Novo Transcriptome Assembly
2.4. Functional Annotation and Gene Ontology Enrichment Analysis
2.5. Inferring Sex-Linked Contigs
2.6. Correcting Mapping Bias
2.7. Estimating the Age of Sex Chromosomes
2.8. Estimating Gene Loss
2.9. Analysis of Expression Level Differences between X and Y Alleles
2.9.1. Allelic Expression Measurement
2.9.2. Analysis of Dosage Compensation in X-Hemizygous Contigs
2.10. Identifying Contigs with Sex-Biased Expression
2.11. Statistics
3. Results
3.1. Sex-Linked Genes Identified by SEX-DETector
3.2. Age of the C. grandis XY System
3.3. Patterns of Y Degeneration
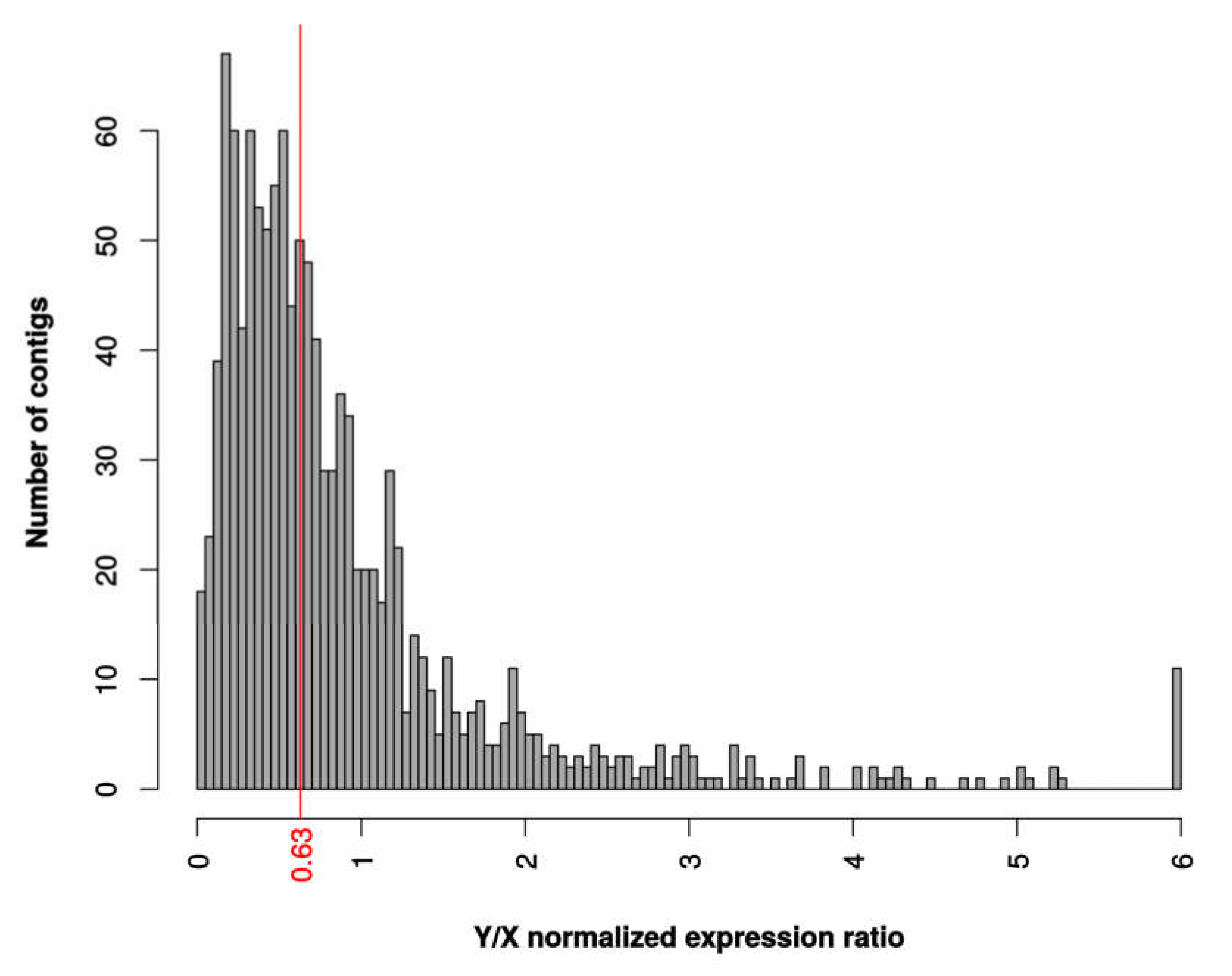
3.4. Patterns of Dosage Compensation
3.5. Genomic Distribution of Sex-Biased Genes
4. Discussion
4.1. Coccinia grandis XY are of Intermediate Age, Similarly to other Highly Heteromorphic Plant Systems
4.2. Coccinia grandis Y Chromosome Degeneration is Moderate, with an Unusually Reduced Y Expression
4.3. Coccinia grandis Exhibit Sex Chromosome Dosage Compensation, a Phenomenon Observed in Several Plant Systems
4.4. Coccinia grandis Sex Chromosomes are Enriched in Sex-Biased Genes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Renner, S.S. The relative and absolute frequencies of angiosperm sexual systems: Dioecy, monoecy, gynodioecy, and an updated online database. Am. J. Bot. 2014, 101, 1588–1596. [Google Scholar] [CrossRef] [PubMed]
- Ming, R.; Bendahmane, A.; Renner, S.S. Sex chromosomes in land plants. Annu. Rev. Plant Biol. 2011, 62, 485–514. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D.; Charlesworth, B.; Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 2005, 95, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Bergero, R.; Charlesworth, D. The evolution of restricted recombination in sex chromosomes. Trends Ecol. Evol. 2009, 24, 94–102. [Google Scholar] [CrossRef]
- Muyle, A.; Shearn, R.; Marais, G.A. The evolution of sex chromosomes and dosage compensation in plants. Genome Biol. Evol. 2017, 9, 627–645. [Google Scholar] [CrossRef]
- Bachtrog, D. Y-chromosome evolution: Emerging insights into processes of Y-chromosome degeneration. Nat. Rev. Genome 2013, 14, 113–124. [Google Scholar] [CrossRef]
- Mahajan, S.; Wei, K.H.-C.; Nalley, M.J.; Gibilisco, L.; Bachtrog, D. De novo assembly of a young Drosophila Y chromosome using single-molecule sequencing and chromatin conformation capture. PLoS Biol. 2018, 16, e2006348. [Google Scholar] [CrossRef]
- Filatov, D.A.; Moneger, F.; Negrutiu, I.; Charlesworth, D. Low variability in a Y-linked plant gene and its implications for Y-chromosome evolution. Nature 2000, 404, 388–390. [Google Scholar] [CrossRef]
- Nicolas, M.; Marais, G.; Hykelova, V.; Janousek, B.; Laporte, V.; Vyskot, B.; Mouchiroud, D.; Negrutiu, I.; Charlesworth, D.; Moneger, F. A gradual process of recombination restriction in the evolutionary history of the sex chromosomes in dioecious plants. PLoS Biol. 2005, 3, e4. [Google Scholar] [CrossRef]
- Bergero, R.; Charlesworth, D.; Filatov, D.A.; Moore, R.C. Defining regions and rearrangements of the Silene latifolia Y chromosome. Genetics 2008, 178, 2045–2053. [Google Scholar] [CrossRef]
- Cermak, T.; Kubat, Z.; Hobza, R.; Koblizkova, A.; Widmer, A.; Macas, J.; Vyskot, B.; Kejnovsky, E. Survey of repetitive sequences in Silene latifolia with respect to their distribution on sex chromosomes. Chromosome Res. 2008, 16, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Marais, G.; Nicolas, M.; Bergero, R.; Chambrier, P.; Kejnovsky, E.; Monéger, F.; Hobza, R.; Widmer, A.; Charlesworth, D. Evidence for degeneration of the Y chromosome in the dioecious plant Silene Latifolia. Curr. Biol. 2008, 18, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Chibalina, M.V.; Filatov, D.A. Plant Y chromosome degeneration is retarded by haploid purifying selection. Curr. Biol. 2011, 21, 1475–1479. [Google Scholar] [CrossRef] [PubMed]
- Bergero, R.; Charlesworth, D. Preservation of the Y transcriptome in a 10-million-year-old plant sex chromosome system. Curr. Biol. 2011, 21, 1470–1474. [Google Scholar] [CrossRef]
- Muyle, A.; Zemp, N.; Deschamps, C.; Mousset, S.; Widmer, A.; Marais, G.A. Rapid de novo evolution of X chromosome dosage compensation in Silene latifolia, a plant with young sex chromosomes. PLoS Biol. 2012, 10, e1001308. [Google Scholar] [CrossRef]
- Papadopulos, A.S.; Chester, M.; Ridout, K.; Filatov, D.A. Rapid Y degeneration and dosage compensation in plant sex chromosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 13021–13026. [Google Scholar] [CrossRef]
- Muyle, A.; Zemp, N.; Fruchard, C.; Cegan, R.; Vrana, J.; Deschamps, C.; Tavares, R.; Hobza, R.; Picard, F.; Widmer, A.; et al. Genomic imprinting mediates dosage compensation in a young plant XY system. Nat. Plants 2018, 4, 677–680. [Google Scholar] [CrossRef]
- Lorenzo, J.L.R.; Hobza, R.; Vyskot, B. DNA methylation and genetic degeneration of the Y chromosome in the dioecious plant Silene latifolia. BMC Gen. 2018, 19, 540. [Google Scholar]
- Gu, L.; Walters, J.R. Evolution of sex chromosome dosage compensation in animals: A beautiful theory, undermined by facts and bedeviled by details. Genome Biol. Evol. 2017, 9, 2461–2476. [Google Scholar] [CrossRef]
- Ercan, S. Mechanisms of X chromosome dosage compensation. J. Gen. 2015, 3, 1. [Google Scholar] [CrossRef]
- Pessia, E.; Makino, T.; Bailly-Bechet, M.; McLysaght, A.; Marais, G.A. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc. Natl. Acad. Sci. USA 2012, 109, 5346–5351. [Google Scholar] [CrossRef]
- Pessia, E.; Engelstädter, J.; Marais, G.A. The evolution of X chromosome inactivation in mammals: The demise of Ohno’s hypothesis? Cell. Mol. Life Sci. 2014, 71, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E. Sex chromosome dosage compensation: Definitely not for everyone. Trends Gen. 2013, 29, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Albritton, S.E.; Kranz, A.-L.; Rao, P.; Kramer, M.; Dieterich, C.; Ercan, S. Sex-biased gene expression and evolution of the x chromosome in nematodes. Genetics 2014, 197, 865–883. [Google Scholar] [CrossRef] [PubMed]
- Veitia, R.A.; Veyrunes, F.; Bottani, S.; Birchler, J.A. X chromosome inactivation and active X upregulation in therian mammals: Facts, questions, and hypotheses. J. Mol. Cell Biol. 2015, 7, 2–11. [Google Scholar] [CrossRef]
- Arnold, A.P.; Itoh, Y.; Melamed, E. A bird’s-eye view of sex chromosome dosage compensation. Annu. Rev. Gen. Hum. Genet. 2008, 9, 109–127. [Google Scholar] [CrossRef]
- Zimmer, F.; Harrison, P.W.; Dessimoz, C.; Mank, J.E. Compensation of dosage-sensitive genes on the chicken Z chromosome. Gen. Biol. Evol. 2016, 8, 1233–1242. [Google Scholar] [CrossRef]
- Bergero, R.; Qiu, S.; Charlesworth, D. Gene loss from a plant sex chromosome system. Curr. Biol. 2015, 25, 1234–1240. [Google Scholar] [CrossRef]
- Blavet, N.; Blavet, H.; Muyle, A.; Kafer, J.; Cegan, R.; Deschamps, C.; Zemp, N.; Mousset, S.; Aubourg, S.; Bergero, R.; et al. Identifying new sex-linked genes through BAC sequencing in the dioecious plant Silene latifolia. BMC Gen. 2015, 16, 546. [Google Scholar] [CrossRef]
- Ohno, S. Sex. Chromosomes and Sex. Linked Genes; Springer: Berlin, Germany, 1967. [Google Scholar]
- Bergero, R. How to solve the gender expression gap. Nat. Plants 2018, 4, 637–638. [Google Scholar] [CrossRef]
- Krasovec, M.; Kazama, Y.; Ishii, K.; Abe, T.; Filatov, D.A. Immediate dosage Compensation is triggered by the deletion of Y-linked genes in Silene latifolia. Curr. Biol. 2019, 29, 2214–2221. [Google Scholar] [CrossRef] [PubMed]
- Siroky, J.; Ruffini Castiglione, M.; Vyskot, B. DNA methylation patterns of Melandrium album chromosomes. Chromosome Res. 1998, 6, 441–446. [Google Scholar] [CrossRef]
- Bačovský, V.; Houben, A.; Kumke, K.; Hobza, R. The distribution of epigenetic histone marks differs between the X and Y chromosomes in Silene latifolia. Planta 2019, 250, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Martin, H.; Carpentier, F.; Gallina, S.; Gode, C.; Schmitt, E.; Muyle, A.; Marais, G.A.B.; Touzet, P. Evolution of young sex chromosomes in two dioecious sister plant species with distinct sex determination systems. Genome Biol. Evol. 2019, 11, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Crowson, D.; Barrett, S.C.H.; Wright, S.I. Purifying and positive selection influence patterns of gene loss and gene expression in the evolution of a plant sex chromosome system. Mol. Biol. Evol. 2017, 34, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Prentout, D.; Razumova, O.; Rhoné, B.; Badouin, H.; Henri, H.; Feng, C.; Käfer, J.; Karlov, G.; Marais, G.A. An efficient RNA-seq-based segregation analysis identifies the sex chromosomes of Cannabis sativa. Genome Res. 2020, 30, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Kumar, L.S.S.; Vishveshwaraiah, S. Sex mechanism in Coccinia indica Wight and Arn. Nature 1952, 170, 330–331. [Google Scholar] [CrossRef]
- Sousa, A.; Fuchs, J.; Renner, S.S. Molecular cytogenetics (FISH, GISH) of Coccinia grandis: A ca. 3 Myr-old species of cucurbitaceae with the largest Y/autosome divergence in flowering plants. Cytogenet. Genome Res. 2013, 139, 107–118. [Google Scholar] [CrossRef]
- Sousa, A.; Bellot, S.; Fuchs, J.; Houben, A.; Renner, S.S. Analysis of transposable elements and organellar DNA in male and female genomes of a species with a huge Y chromosome reveals distinct Y centromeres. Plant J. 2016, 88, 387–396. [Google Scholar] [CrossRef]
- Sousa, A.; Fuchs, J.; Renner, S.S. Cytogenetic comparison of heteromorphic and homomorphic sex chromosomes in Coccinia (Cucurbitaceae) points to sex chromosome turnover. Chromosome Res. 2017, 25, 191–200. [Google Scholar] [CrossRef]
- Holstein, N.; Renner, S.S. A dated phylogeny and collection records reveal repeated biome shifts in the African genus Coccinia (Cucurbitaceae). BMC Evol. Biol. 2011, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- Devani, R.S.; Sinha, S.; Banerjee, J.; Sinha, R.K.; Bendahmane, A.; Banerjee, A.K. De novo transcriptome assembly from flower buds of dioecious, gynomonoecious and chemically masculinized female Coccinia grandis reveals genes associated with sex expression and modification. BMC Plant Biol. 2017, 17, 241. [Google Scholar] [CrossRef]
- Mohanty, J.N.; Nayak, S.; Jha, S.; Joshi, R.K. Transcriptome profiling of the floral buds and discovery of genes related to sex-differentiation in the dioecious cucurbit Coccinia grandis (L.) Voigt. Gene 2017, 626, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, H.; Heibl, C.; Renner, S.S. Gourds afloat: A dated phylogeny reveals an Asian origin of the gourd family (Cucurbitaceae) and numerous oversea dispersal events. Proc. R. Soc. B Biol. Sci. 2009, 276, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef]
- Garcia-Mas, J.; Benjak, A.; Sanseverino, W.; Bourgeois, M.; Mir, G.; González, V.M.; Hénaff, E.; Câmara, F.; Cozzuto, L.; Lowy, E.; et al. The genome of melon (Cucumis melo L.). Proc. Natl. Acad. Sci. USA 2012, 109, 11872–11877. [Google Scholar] [CrossRef]
- Ruggieri, V.; Alexiou, K.G.; Morata, J.; Argyris, J.; Pujol, M.; Yano, R.; Nonaka, S.; Ezura, H.; Latrasse, D.; Boualem, A.; et al. An improved assembly and annotation of the melon (Cucumis melo L.) reference genome. Sci Rep. 2018, 8, 8088. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Huang, W.; Xu, Y.; Zhou, Q.; Wang, S.; Ruan, J.; Huang, S.; Zhang, Z. A chromosome-scale genome assembly of cucumber (Cucumis sativus L.). GigaScience 2019, 8. [Google Scholar] [CrossRef]
- Muyle, A.; Kafer, J.; Zemp, N.; Mousset, S.; Picard, F.; Marais, G.A. SEX-DETector: A probabilistic approach to study sex chromosomes in non-model organisms. Genome Biol. Evol. 2016, 8, 2530–2543. [Google Scholar] [CrossRef]
- Hough, J.; Hollister, J.D.; Wang, W.; Barrett, S.C.; Wright, S.I. Genetic degeneration of old and young Y chromosomes in the flowering plant Rumex hastatulus. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef]
- Michalovova, M.; Kubat, Z.; Hobza, R.; Vyskot, B.; Kejnovsky, E. Fully automated pipeline for detection of sex linked genes using RNA-Seq data. BMC Bioinform. 2015, 16, 78. [Google Scholar] [CrossRef] [PubMed]
- Zemp, N.; Tavares, R.; Muyle, A.; Charlesworth, D.; Marais, G.A.; Widmer, A. Evolution of sex-biased gene expression in a dioecious plant. Nat. Plants 2016, 2, 16168. [Google Scholar] [CrossRef] [PubMed]
- Veltsos, P.; Ridout, K.E.; Toups, M.A.; Gonzalez-Martinez, S.C.; Muyle, A.; Emery, O.; Rastas, P.; Hudzieczek, V.; Hobza, R.; Vyskot, B.; et al. Early sex-chromosome evolution in the diploid dioecious plant Mercurialis annua. Genetics 2019. [Google Scholar] [CrossRef] [PubMed]
- Badouin, H.; Velt, A.; Gindraud, F.; Flutre, T.; Dumas, V.; Vautrin, S.; Marande, W.; Corbi, J.; Sallet, E.; Ganofsky, J.; et al. The wild grape genome sequence provides insights into the transition from dioecy to hermaphroditism during grape domestication. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ghadge, A.G.; Karmakar, K.; Devani, R.S.; Banerjee, J.; Mohanasundaram, B.; Sinha, R.K.; Sinha, S.; Banerjee, A.K. Flower development, pollen fertility and sex expression analyses of three sexual phenotypes of Coccinia grandis. BMC Plant Biol. 2014, 14, 325. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Smeds, L.; Künstner, A. ConDeTri-a content dependent read trimmer for Illumina data. PLoS ONE 2011, 6, e26314. [Google Scholar] [CrossRef]
- Aronesty, E. Ea-utils: “Command-Line Tools for Processing Biological Sequencing Data”. 2011. Available online: https://github.com/ExpressionAnalysis/ea-utils (accessed on 12 July 2017).
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Schmieder, R.; Lim, Y.W.; Edwards, R. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 2012, 28, 433–435. [Google Scholar] [CrossRef]
- Huang, X.; Madan, A. CAP3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. goseq: Gene Ontology testing for RNA-seq datasets. R Bioconductor 2012, 8, 1–25. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Gayral, P.; Melo-Ferreira, J.; Glemin, S.; Bierne, N.; Carneiro, M.; Nabholz, B.; Lourenco, J.M.; Alves, P.C.; Ballenghien, M.; Faivre, N.; et al. Reference-free population genomics from next-generation transcriptome data and the vertebrate-invertebrate gap. PLoS Genet. 2013, 9, e1003457. [Google Scholar] [CrossRef]
- Tsagkogeorga, G.; Cahais, V.; Galtier, N. The population genomics of a fast evolver: High levels of diversity, functional constraint, and molecular adaptation in the tunicate Ciona intestinalis. Genome Biol. Evol. 2012, 4, 740–749. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Koch, M.; Haubold, B.; Mitchell-Olds, T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis and related genera (Brassicaceae). Mol. Biol. Evol. 2000, 17, 1483–1498. [Google Scholar] [CrossRef]
- Ossowski, S.; Schneeberger, K.; Lucas-Lledó, J.I.; Warthmann, N.; Clark, R.M.; Shaw, R.G.; Lynch, M. The rate and molecular spectrum of spontaneous mutations in Arabidopsis Thaliana. Science 2010, 327, 92–94. [Google Scholar] [CrossRef]
- Beilstein, M.A.; Nagalingum, N.S.; Clements, M.D.; Manchester, S.R.; Mathews, S. Dated molecular phylogenies indicate a Miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 18724–18728. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Mullon, C.; Wright, A.E.; Reuter, M.; Pomiankowski, A.; Mank, J.E. Evolution of dosage compensation under sexual selection differs between X and Z chromosomes. Nat. Commun. 2015, 6, 7720. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McCarthy, D.J.; Chen, Y.; Smyth, G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucl. Acids Res. 2012, 40, 4288–4297. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015; Available online: http/www.R-project.org (accessed on 30 November 2017).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Krasovec, M.; Chester, M.; Ridout, K.; Filatov, D.A. The Mutation Rate and the Age of the Sex Chromosomes in Silene latifolia. Curr. Biol. 2018, 28, 1832–1838. [Google Scholar] [CrossRef]
- Rautenberg, A.; Sloan, D.B.; Alden, V.; Oxelman, B. Phylogenetic relationships of Silene multinervia and Silene section conoimorpha (Caryophyllaceae). Syst. Bot. 2012, 37, 226–237. [Google Scholar] [CrossRef]
- Hollister, J.D.; Smith, L.M.; Guo, Y.L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef]
- Gorelick, R. Evolution of dioecy and sex chromosomes via methylation driving Muller’s ratchet. Biol. J. Linn. Soc. 2003, 80, 353–368. [Google Scholar] [CrossRef]
- Birchler, J.A. A study of enzyme activities in a dosage series of the long arm of chromosome one in maize. Genetics 1979, 92, 1211–1229. [Google Scholar] [PubMed]
- Birchler, J.A. The genetic basis of dosage compensation of alcohol dehydrogenase-1 in maize. Genetics 1981, 97, 625–637. [Google Scholar]
- Birchler, J.A.; Newton, K.J. Modulation of protein levels in chromosomal dosage series of maize: The biochemical basis of aneuploid syndromes. Genetics 1981, 99, 247–266. [Google Scholar] [PubMed]
- Birchler, J.A.; Hiebert, J.C.; Paigen, K. Analysis of autosomal dosage compensation involving the alcohol dehydrogenase locus in Drosophila melanogaster. Genetics 1990, 124, 679–686. [Google Scholar] [PubMed]
- Guo, M.; Birchler, J.A. Tran-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 1994, 266, 1999–2002. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Johnson, A.F.; Donohue, R.C.; Li, J.; Cheng, J.; Birchler, J.A. Dosage compensation and inverse effects in triple X metafemales of Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 7383–7388. [Google Scholar] [CrossRef]
- Sun, L.; Johnson, A.F.; Li, J.; Lambdin, A.S.; Cheng, J.; Birchler, J.A. Differential effect of aneuploidy on the X chromosome and genes with sex-biased expression in Drosophila. Proc. Natl. Acad. Sci. USA 2013, 110, 16514–16519. [Google Scholar] [CrossRef]
- Hou, J.; Shi, X.; Chen, C.; Islam, M.S.; Johnson, A.F.; Kanno, T.; Huettel, B.; Yen, M.R.; Hsu, F.M.; Ji, T.; et al. Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proc. Natl Acad. Sci. USA 2018, 115, E11321–E11330. [Google Scholar] [CrossRef]
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef]
- Hurst, L.D.; Ghanbarian, A.T.; Forrest, A.R.; Huminiecki, L. The constrained maximal expression level owing to haploidy shapes gene content on the mammalian X chromosome. PLoS Biol. 2015, 13, e1002315. [Google Scholar] [CrossRef] [PubMed]
- Harkess, A.; Mercati, F.; Shan, H.Y.; Sunseri, F.; Falavigna, A.; Leebens-Mack, J. Sex-biased gene expression in dioecious garden asparagus (Asparagus officinalis). New Phytol. 2015, 207, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Cossard, G.G.; Toups, M.A.; Pannell, J.R. Sexual dimorphism and rapid turnover in gene expression in pre-reproductive seedlings of a dioecious herb. Ann. Bot. 2019, 123, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Darolti, I.; Wright, A.E.; Pucholt, P.; Berlin, S.; Mank, J.E. Slow evolution of sex-biased genes in the reproductive tissue of the dioecious plant Salix viminalis. Mol. Ecol. 2018, 27, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, B.J.; Wang, L.; Tiffin, P.; Wu, Z.; Olson, M.S. Sex-biased gene expression in flowers, but not leaves, reveals secondary sexual dimorphism in Populus balsamifera. New Phytol. 2019, 221, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Muyle, A. How different is the evolution of sex-biased gene expression between plants and animals? A commentary on: ‘Sexual dimorphism and rapid turnover in gene expression in pre-reproductive seedlings of a dioecious herb’. Ann. Bot. 2019, 123, iv–v. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Full Transcriptome | Longest ORF per Isoform | |
|---|---|---|
| Total contigs | 128,904 | 82,699 |
| Total assembled bases (bp) | 103,275,123 | 27,290,670 |
| Median contig length | 552 | 836 |
| Average contig length | 801.18 | 836.83 |
| Maximum contig length | 16,296 | 16,296 |
| Minimum contig length | 297 | 297 |
| N50 | 1029 | 1086 |
| Total contigs longer than 1kb | 30,795 | 21,587 |
| GC content (%) | 42.96 | 42.96 |
| SEX-DETector with BWA Mapping | SEX-DETector with GSNAP SNP-Tolerant Mapping | |
| Contigs in final assembly | 82,699 | 82,699 |
| Contigs with enough coverage to be studied | 82,689 | 70,298 |
| Contigs with enough informative SNPs to compute a segregation probability | 4320 | 3801 |
| Contigs assigned to an autosomal segregation type | 2889 | 3706 |
| Contigs assigned to a X-Y segregation type | 1239 | 1196 |
| Contigs assigned to a X-hemizygous segregation type | 192 | 168 |
| Molecular Clocks | Age Estimates of the Sex Chromosomes, with dS max = 0.17 | Age Estimates of the Sex Chromosomes, with dS max = 0.13 |
|---|---|---|
| From [70], calibrated with an assumed divergence time of Barbarea and Cardamine of 6.0 My | 11.3 | 8.7 |
| From [71], generation time = 1 year, assumed for Arabidopsis thaliana | 12.1 | 9.3 |
| From [71], generation time = 1.5 year | 18.2 | 13.9 |
| From [72], calibrated with six Brassicales fossils | 34.7 | 26.5 |
| From [71], generation time = 5.5 year | 66.8 | 51.1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fruchard, C.; Badouin, H.; Latrasse, D.; Devani, R.S.; Muyle, A.; Rhoné, B.; Renner, S.S.; Banerjee, A.K.; Bendahmane, A.; Marais, G.A.B. Evidence for Dosage Compensation in Coccinia grandis, a Plant with a Highly Heteromorphic XY System. Genes 2020, 11, 787. https://doi.org/10.3390/genes11070787
Fruchard C, Badouin H, Latrasse D, Devani RS, Muyle A, Rhoné B, Renner SS, Banerjee AK, Bendahmane A, Marais GAB. Evidence for Dosage Compensation in Coccinia grandis, a Plant with a Highly Heteromorphic XY System. Genes. 2020; 11(7):787. https://doi.org/10.3390/genes11070787
Chicago/Turabian StyleFruchard, Cécile, Hélène Badouin, David Latrasse, Ravi S. Devani, Aline Muyle, Bénédicte Rhoné, Susanne S. Renner, Anjan K. Banerjee, Abdelhafid Bendahmane, and Gabriel A. B. Marais. 2020. "Evidence for Dosage Compensation in Coccinia grandis, a Plant with a Highly Heteromorphic XY System" Genes 11, no. 7: 787. https://doi.org/10.3390/genes11070787
APA StyleFruchard, C., Badouin, H., Latrasse, D., Devani, R. S., Muyle, A., Rhoné, B., Renner, S. S., Banerjee, A. K., Bendahmane, A., & Marais, G. A. B. (2020). Evidence for Dosage Compensation in Coccinia grandis, a Plant with a Highly Heteromorphic XY System. Genes, 11(7), 787. https://doi.org/10.3390/genes11070787