Identification of QTNs and Their Candidate Genes for 100-Seed Weight in Soybean (Glycine max L.) Using Multi-Locus Genome-Wide Association Studies



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Statistical Analysis and Heritability Estimation
2.3. Population Structure Analysis and Genome-Wide Association Studies
2.4. Elite Allele Analysis
2.5. Prediction of Candidate Genes for 100-Seed Weight
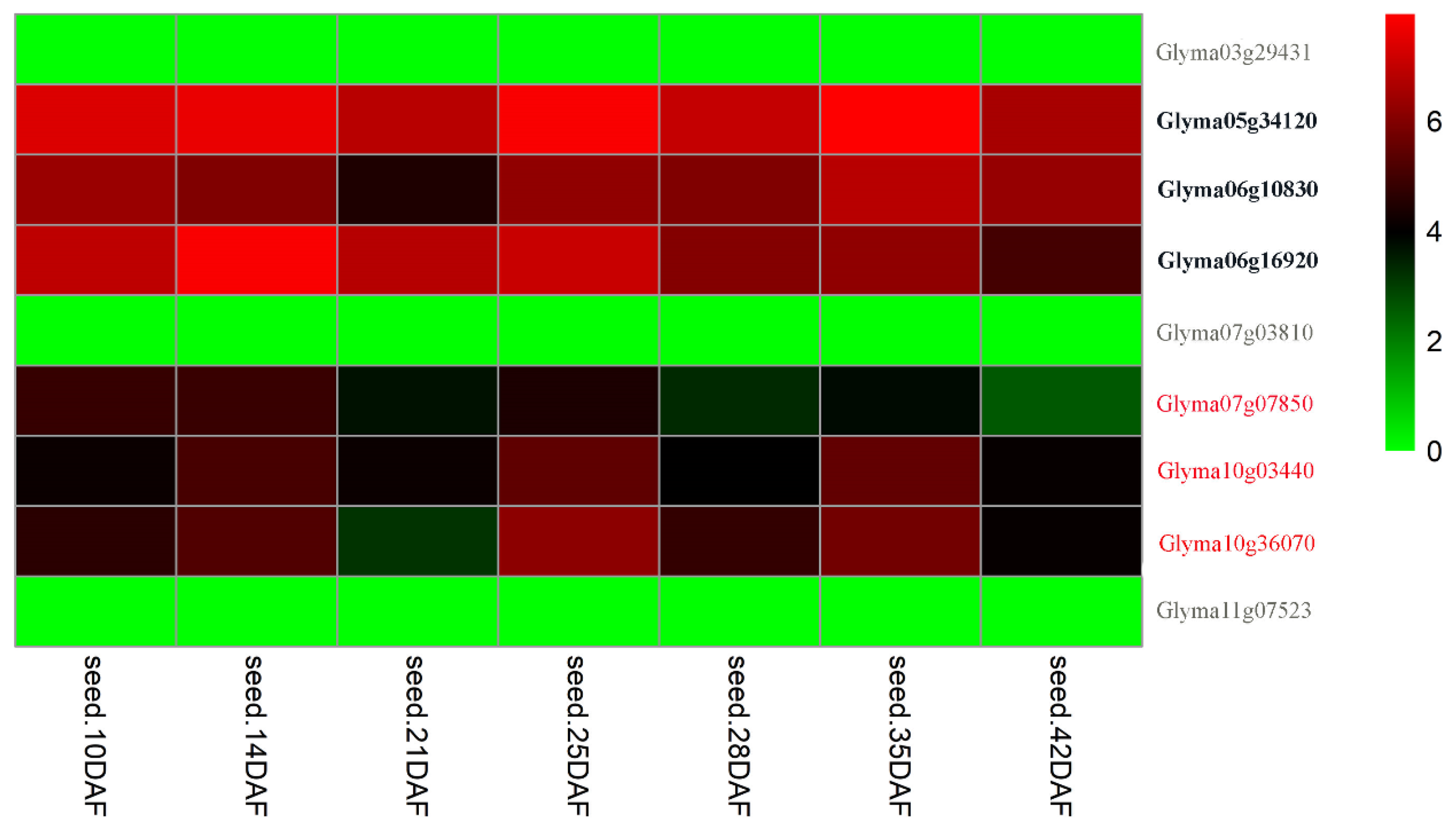
2.5.1. Gene Expression Level Analysis
2.5.2. Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
3. Results
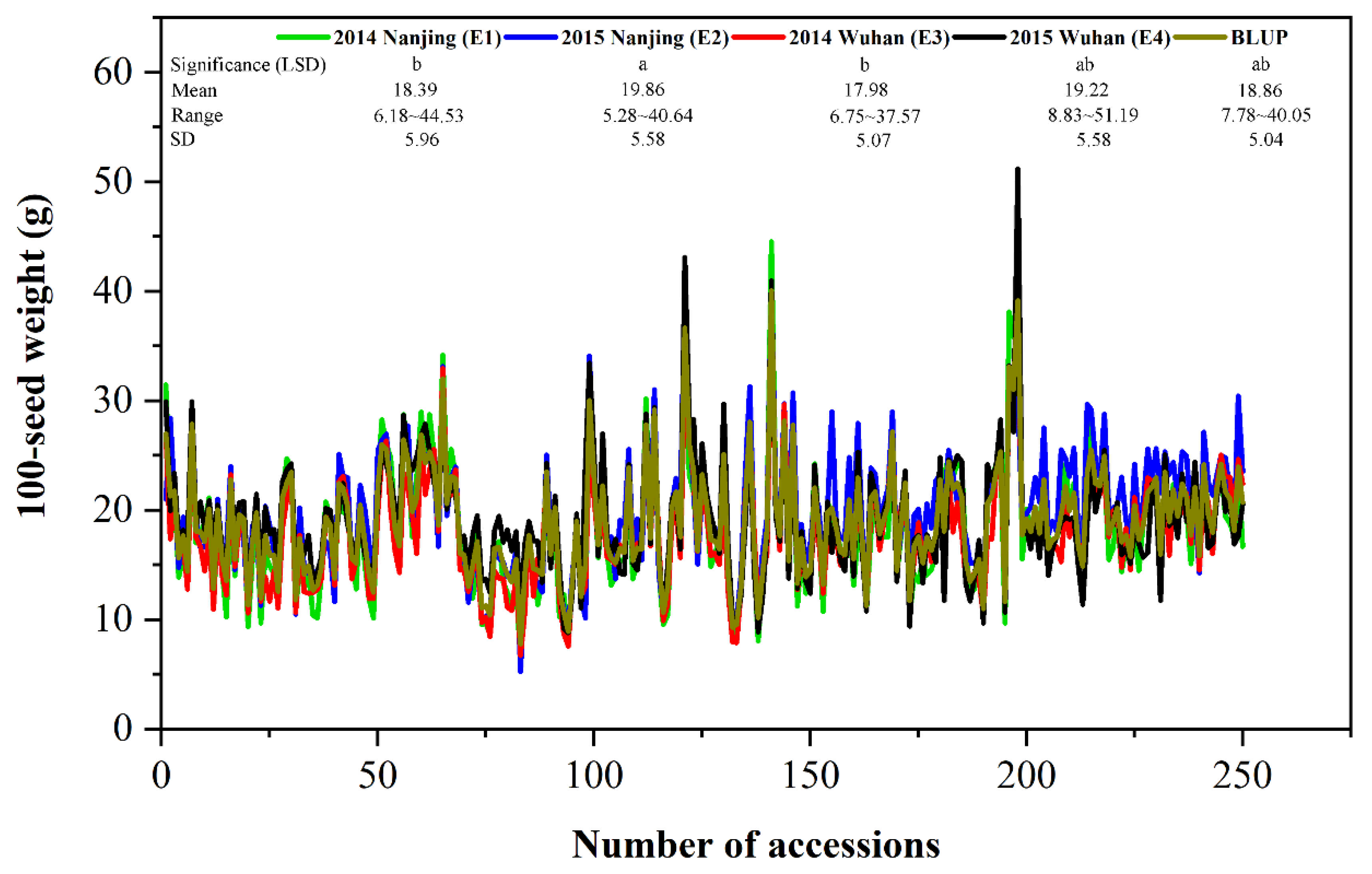
3.1. Phenotype Variation of 100-Seed Weight
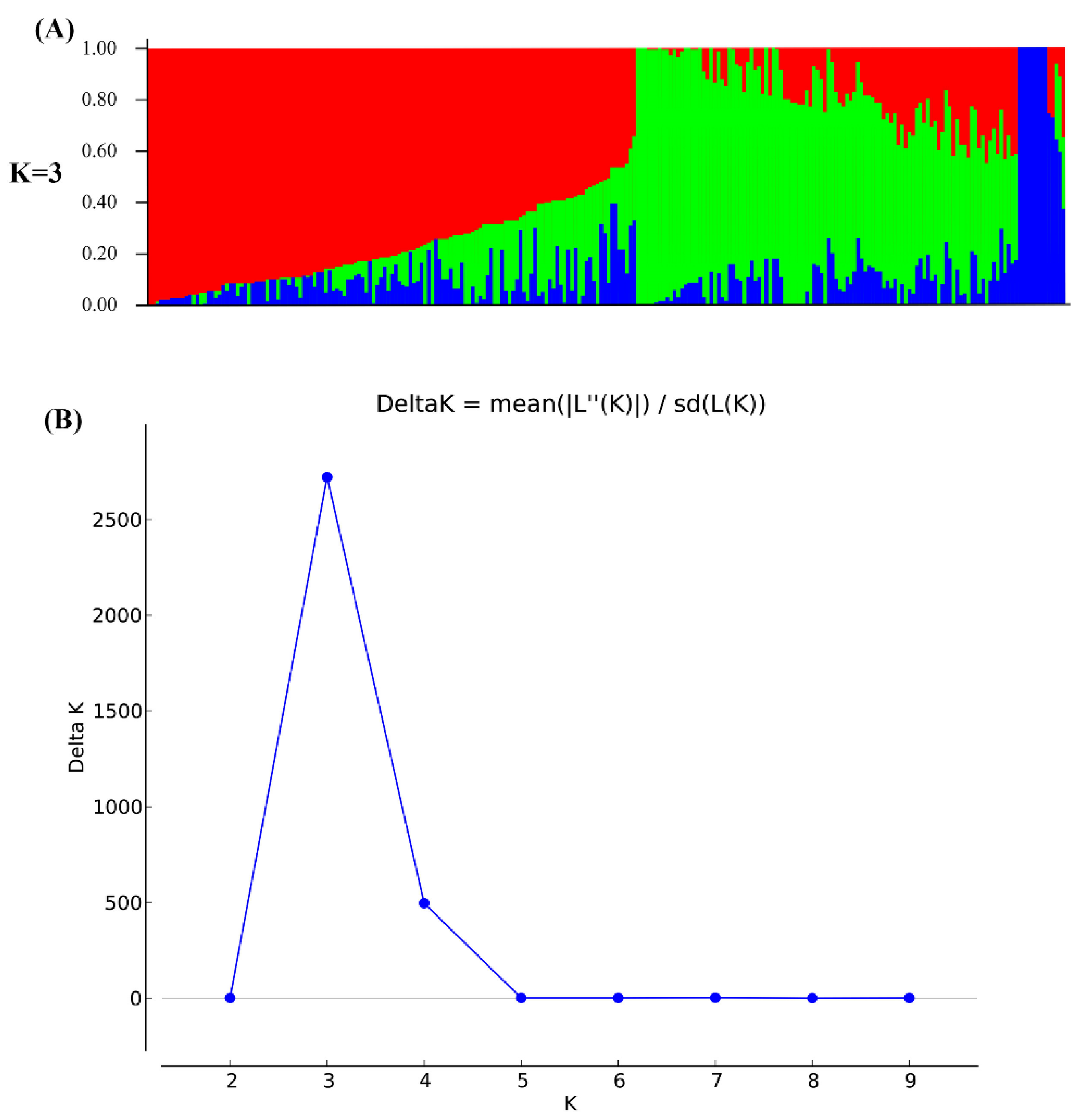
3.2. Population Structure Analysis and Genome-Wide Association Studies
3.3. Stable QTNs for 100-SW in Soybean
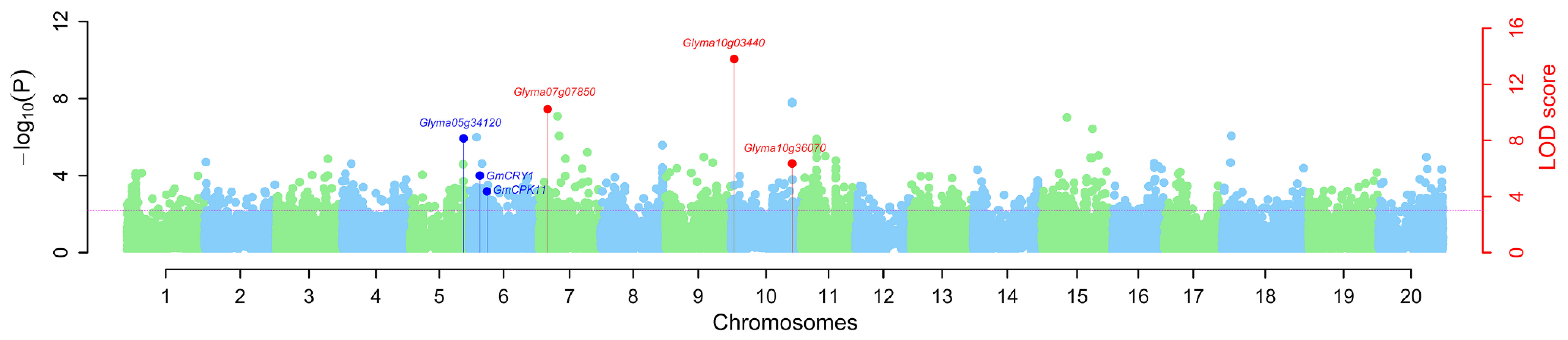
3.4. Validation of Stable QTNs for 100-SW in Soybean
3.5. Prediction of the Best Parental Combinations for 100-Seed Weight in Two Directions
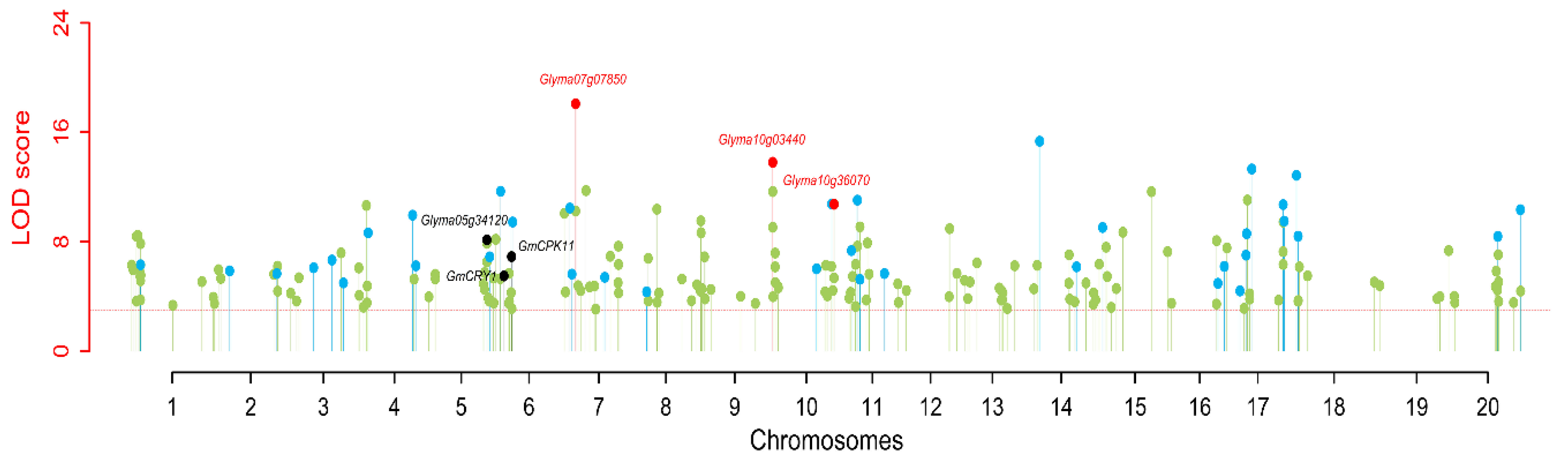
3.6. Candidate Genes Underlying the Stable QTNs for 100-SW in Soybean
4. Discussion
4.1. Comparison of Stable QTNs in This Study with Previously Reported QTLs
4.2. Reliability of QTNs and Application of SW Increasing Allele in Soybean Breeding
4.3. Candidate Genes Underlying Stable QTNs for Seed Weight
4.4. Statistical Power of Multi-Locus GWAS Approaches
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.; Yang, G.; Wong, F.L.; Li, M.W.; He, W.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 2010, 42, 1053–1059. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.N.; Li, G.J.; Wang, X.; Cheng, L.G.; Zhang, Y.M. Mapping quantitative trait loci for seed size traits in soybean (Glycine max L. Merr.). Theor. Appl. Genet. 2011, 122, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.S. Food Use of Whole Soybeans. In Soybeans: Chemistry, Production, Processing, and Utilization; AOCS Press: Urbana, IL, USA, 2008; pp. 441–481. [Google Scholar]
- Chen, Y.; Nelson, R.L. Genetic Variation and Relationships among Cultivated, Wild, and Semiwild Soybean. Crop Sci. 2004, 44, 316–325. [Google Scholar] [CrossRef]
- Nawaz, M.A.; Yang, S.H.; Chung, G. Wild Soybeans: An Opportunistic Resource for Soybean Improvement. In Rediscovery of Landraces as a Resource for the Future, 1st ed.; Oscar, G., Ed.; IntechOpen: Rijeka, Croatia, 2018. [Google Scholar]
- Nawaz, M.A.; Rehman, H.M.; Baloch, F.S.; Ijaz, B.; Ali, M.A.; Khan, I.A.; Lee, J.D.; Chung, G.; Yang, S.H. Genome and transcriptome-wide analyses of cellulose synthase gene superfamily in soybean. J. Plant Physiol. 2017, 215, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, X.; Zhu, L.; Bu, Y.; Wang, X.; Zhang, X.; Zhou, Y.; Wang, X.; Guo, N.; Qiu, L.; et al. Genome-wide association study of four yield-related traits at the R6 stage in soybean. BMC Genet. 2019, 20, 1–15. [Google Scholar] [CrossRef]
- Zhao, X.; Dong, H.; Chang, H.; Zhao, J.; Teng, W.; Qiu, L.; Li, W.; Han, Y. Genome wide association mapping and candidate gene analysis for hundred seed weight in soybean [Glycine max (L.) Merrill]. BMC Genom. 2019, 20, 648. [Google Scholar] [CrossRef]
- Zhou, Z.; Lakhssassi, N.; Cullen, M.A.; El Baz, A.; Vuong, T.D.; Nguyen, H.T.; Meksem, K. Assessment of phenotypic variations and correlation among seed composition traits in mutagenized soybean populations. Genes 2019, 10, 975. [Google Scholar] [CrossRef]
- Russell, J.S. Soybeans: Improvement, production, and uses. Field Crops Res. 1988, 18, 299–300. [Google Scholar] [CrossRef]
- Agarwal, M.; Shrivastava, N.; Padh, H. Advances in molecular marker techniques and their applications in plant sciences. Plant Cell Rep. 2008, 27, 617–631. [Google Scholar] [CrossRef]
- Hoeck, J.A.; Fehr, W.R.; Shoemaker, R.C.; Welke, G.A.; Johnson, S.L.; Cianzio, S.R. Molecular marker analysis of seed size in soybean. Crop Sci. 2003, 43, 68–74. [Google Scholar] [CrossRef]
- Mian, M.A.R.; Bailey, M.A.; Tamulonis, J.P.; Shipe, E.R.; Carter, T.E.; Parrott, W.A.; Ashley, D.A.; Hussey, R.S.; Boerma, H.R. Molecular markers associated with seed weight in two soybean populations. Theor. Appl. Genet. 1996, 93, 1011–1016. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, D.; Zhu, D.; Li, H.; Li, X.; Teng, W.; Li, W. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theor. Appl. Genet. 2012, 125, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.T.; Niu, Y.; Zhang, J.; Bu, S.H.; Zhang, H.Z.; Geng, Q.C.; Feng, J.Y.; Zhang, Y.M. Fine mapping of quantitative trait loci for seed size traits in soybean. Mol. Breed. 2014, 34, 2165–2178. [Google Scholar] [CrossRef]
- Mansur, L.M.; Orf, J.H.; Chase, K.; Jarvik, T.; Cregan, P.B.; Lark, K.G. Genetic mapping of agronomic traits using recombinant inbred lines of soybean. Crop Sci. 1996, 36, 1327–1336. [Google Scholar] [CrossRef]
- Teng, W.; Han, Y.; Du, Y.; Sun, D.; Zhang, Z.; Qiu, L.; Sun, G.; Li, W. QTL analyses of seed weight during the development of soybean (Glycine max L. Merr.). Heredity 2009, 102, 372–380. [Google Scholar] [CrossRef]
- Niu, Y.; Xu, Y.; Liu, X.F.; Yang, S.X.; Wei, S.P.; Xie, F.T.; Zhang, Y.M. Association mapping for seed size and shape traits in soybean cultivars. Mol. Breed. 2013, 31, 785–794. [Google Scholar] [CrossRef]
- Thomson, M.J. High-Throughput SNP Genotyping to Accelerate Crop Improvement. Plant Breed. Biotechnol. 2014, 2, 195–212. [Google Scholar] [CrossRef]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Ron, M.; Weller, J.I. From QTL to QTN identification in livestock—Winning by points rather than knock-out: A review. Anim. Genet. 2007, 38, 429–439. [Google Scholar] [CrossRef]
- Wang, S.B.; Feng, J.Y.; Ren, W.L.; Huang, B.; Zhou, L.; Wen, Y.J.; Zhang, J.; Dunwell, J.M.; Xu, S.; Zhang, Y.M. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci. Rep. 2016, 6, 19444. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. Editorial: The applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits. Front. Plant Sci. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Hao, D.; Cheng, H.; Yin, Z.; Cui, S.; Zhang, D.; Wang, H.; Yu, D. Identification of single nucleotide polymorphisms and haplotypes associated with yield and yield components in soybean (Glycine max) landraces across multiple environments. Theor. Appl. Genet. 2012, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, S.B.; Jian, J.; Geng, Q.C.; Wen, J.; Song, Q.; Wu, Z.; Li, G.J.; Liu, Y.Q.; Dunwell, J.M.; et al. Identification of domestication-related loci associated with flowering time and seed size in soybean with the RAD-seq genotyping method. Sci. Rep. 2015, 5, 9350. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Song, Q.; Cregan, P.B.; Jiang, G.L. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean (Glycine max). Theor. Appl. Genet. 2016, 129, 117–130. [Google Scholar] [CrossRef]
- Yan, L.; Hofmann, N.; Li, S.; Ferreira, M.E.; Song, B.; Jiang, G.; Ren, S.; Quigley, C.; Fickus, E.; Cregan, P.; et al. Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses. BMC Genom. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Zhao, X.; Wang, J.; Teng, W.; Qiu, L.; Han, Y.; Li, W. Identification of the genomic region underlying seed weight per plant in soybean (Glycine max L. Merr.) via high-throughput single-nucleotide polymorphisms and a genome-wide association study. Front. Plant Sci. 2018, 9, 1392. [Google Scholar] [CrossRef]
- Assefa, T.; Otyama, P.I.; Brown, A.V.; Kalberer, S.R.; Kulkarni, R.S.; Cannon, S.B. Genome-wide associations and epistatic interactions for internode number, plant height, seed weight and seed yield in soybean. BMC Genom. 2019, 20, 527. [Google Scholar] [CrossRef]
- Hu, D.; Zhang, H.; Du, Q.; Hu, Z.; Yang, Z.; Li, X.; Wang, J.; Huang, F.; Yu, D.; Wang, H.; et al. Genetic dissection of yield-related traits via genome-wide association analysis across multiple environments in wild soybean (Glycine soja Sieb. and Zucc.). Planta 2020, 251, 39. [Google Scholar] [CrossRef]
- Jofuku, K.D.; Omidyar, P.K.; Gee, Z.; Okamuro, J.K. Control of seed mass and seed yield by the floral homeotic gene APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3117–3122. [Google Scholar] [CrossRef]
- Ohto, M.A.; Fischer, R.L.; Goldberg, R.B.; Nakamura, K.; Harada, J.J. Control of seed mass by APETALA2. Proc. Natl. Acad. Sci. USA 2005, 102, 3123–3128. [Google Scholar] [CrossRef]
- Schruff, M.C.; Spielman, M.; Tiwari, S.; Adams, S.; Fenby, N.; Scott, R.J. The AUXIN RESPONSE FACTOR 2 gene of Arabidopsis links auxin signalling, cell division, and the size of seeds and other organs. Development 2006, 133, 251–261. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, X.; Kang, X.; Zhao, X.; Zhang, X.; Ni, M. Short Hypocotyl Under Blue1 associates with Miniseed3 and Haiku2 promoters in vivo to regulate Arabidopsis seed development. Plant Cell 2009, 21, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Shantharaj, D.; Kang, X.; Ni, M. Transcriptional and hormonal signaling control of Arabidopsis seed development. Curr. Opin. Plant Biol. 2010, 13, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, Q.T.; Xiong, Q.; Li, W.; Bi, Y.D.; Lai, Y.C.; Liu, X.L.; Man, W.Q.; Zhang, W.K.; Ma, B.; et al. The transcriptomic signature of developing soybean seeds reveals the genetic basis of seed trait adaptation during domestication. Plant J. 2016, 86, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Xiong, Q.; Cheng, T.; Li, Q.T.; Liu, X.L.; Bi, Y.D.; Li, W.; Zhang, W.K.; Ma, B.; Lai, Y.C.; et al. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight. Mol. Plant 2017, 10, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chu, S.; Zhang, H.; Zhu, Y.; Cheng, H.; Yu, D. Development and application of a novel genome-wide SNP array reveals domestication history in soybean. Sci. Rep. 2016, 6, 20728. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Li, W.; Jiang, H.; Wang, Y.; Gao, H.; Liu, M.; Chen, Q.; Lai, Y.; He, C. Differential expression of a WRKY gene between wild and cultivated soybeans correlates to seed size. J. Exp. Bot. 2017, 68, 2717–2729. [Google Scholar] [CrossRef]
- Yang, Z.; Xin, D.; Liu, C.; Jiang, H.; Han, X.; Sun, Y.; Qi, Z.; Hu, G.; Chen, Q. Identification of QTLs for seed and pod traits in soybean and analysis for additive effects and epistatic effects of QTLs among multiple environments. Mol. Genet. Genom. 2013, 288, 651–667. [Google Scholar] [CrossRef]
- Di, S.; Yan, F.; Rodas, F.R.; Rodriguez, T.O.; Murai, Y.; Iwashina, T.; Sugawara, S.; Mori, T.; Nakabayashi, R.; Yonekura-Sakakibara, K.; et al. Linkage mapping, molecular cloning and functional analysis of soybean gene Fg3 encoding flavonol 3-O-glucoside/galactoside (1 → 2) glucosyltransferase. BMC Plant Biol. 2015, 15, 126. [Google Scholar] [CrossRef]
- Zhang, Y.; He, J.; Wang, Y.; Xing, G.; Zhao, J.; Li, Y.; Yang, S.; Palmer, R.G.; Zhao, T.; Gai, J. Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes. J. Exp. Bot. 2015, 66, 6311–6325. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, W.; He, Q.; Xiang, S.; Tian, D.; Zhao, T.; Gai, J. Identifying a wild allele conferring small seed size, high protein content and low oil content using chromosome segment substitution lines in soybean. Theor. Appl. Genet. 2019, 132, 2793–2807. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Jiang, Y.; Wang, Z.; Gou, Z.; Lyu, J.; Li, W.; Yu, Y.; Shu, L.; Zhao, Y.; Ma, Y.; et al. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean. Nat. Biotechnol. 2015, 33, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Luo, L.; Zuo, J.-F.; Yang, L.; Zhang, L.; Guang, X.; Niu, Y.; Jian, J.; Geng, Q.-C.; Liang, L.; et al. Identification and validation of candidate genes associated with domesticated and improved traits in soybean. Plant Genome 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Ma, Y.; Wu, S.; Liu, Z.; Wang, Z.; Yang, R.; Hu, G.; Zhou, Z.; Yu, H.; Zhang, M.; et al. Genome-wide association studies dissect the genetic networks underlying agronomical traits in soybean. Genome Biol. 2017, 18, 161. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, F.; Zhou, Y. The application of multi-locus GWAS for the detection of salt-tolerance loci in rice. Front. Plant Sci. 2018, 9, 1464. [Google Scholar] [CrossRef]
- Ma, L.; Liu, M.; Yan, Y.; Qing, C.; Zhang, X.; Zhang, Y.; Long, Y.; Wang, L.; Pan, L.; Zou, C.; et al. Genetic dissection of maize embryonic callus regenerative capacity using multi-locus genome-wide association studies. Front. Plant Sci. 2018, 9, 561. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Jia, Z.; Dunwell, J.M. (Eds.) The Applications of New Multi-Locus GWAS Methodologies in the Genetic Dissection of Complex Traits; Frontiers Media: Lausanne, Switzerland, 2019. [Google Scholar]
- Bates, D.; Mächler, M.; Bolker, B.M.; Walker, S.C. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Xu, S. Mapping quantitative trait loci by controlling polygenic background effects. Genetics 2013, 195, 1209–1222. [Google Scholar] [CrossRef]
- Ryoo, H.; Lee, C. Underestimation of heritability using a mixed model with a polygenic covariance structure in a genome-wide association study for complex traits. Eur. J. Hum. Genet. 2014, 22, 851–854. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software Structure: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [PubMed]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Zhang, J.; Feng, J.Y.; Ni, Y.L.; Wen, Y.J.; Niu, Y.; Tamba, C.L.; Yue, C.; Song, Q.; Zhang, Y.M. PLARmEB: Integration of least angle regression with empirical Bayes for multilocus genome-wide association studies. Heredity 2017, 118, 517–524. [Google Scholar] [CrossRef]
- Tamba, C.L.; Ni, Y.L.; Zhang, Y.M. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies. PLoS Comput. Biol. 2017, 13, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tamba, C.L.; Zhang, Y.M. A fast mrMLM algorithm for multi-locus genome-wide association studies. bioRxiv 2018, 341784. [Google Scholar] [CrossRef]
- Ren, W.L.; Wen, Y.J.; Dunwell, J.M.; Zhang, Y.M. PKWmEB: Integration of Kruskal-Wallis test with empirical Bayes under polygenic background control for multi-locus genome-wide association study. Heredity 2018, 120, 208–218. [Google Scholar] [CrossRef]
- Li, D.; Zhao, X.; Han, Y.; Li, W.; Xie, F. Genome-wide association mapping for seed protein and oil contents using a large panel of soybean accessions. Genomics 2019, 111, 90–95. [Google Scholar] [CrossRef]
- Jones, S.I.; Vodkin, L.O. Using RNA-seq to profile soybean seed development from fertilization to maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef]
- Severin, A.J.; Woody, J.L.; Bolon, Y.T.; Joseph, B.; Diers, B.W.; Farmer, A.D.; Muehlbauer, G.J.; Nelson, R.T.; Grant, D.; Specht, J.E.; et al. RNA-seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010, 10, 160. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Song, Q.; Jia, G.; Zhu, Y.; Grant, D.; Nelson, R.T.; Hwang, E.Y.; Hyten, D.L.; Cregan, P.B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_1.0) in soybean. Crop Sci. 2010, 50, 1950–1960. [Google Scholar] [CrossRef]
- Aghamirzaie, D.; Batra, D.; Heath, L.S.; Schneider, A.; Grene, R.; Collakova, E. Transcriptome-wide functional characterization reveals novel relationships among differentially expressed transcripts in developing soybean embryos. BMC Genom. 2015, 16, 928. [Google Scholar] [CrossRef] [PubMed]
- Sreeramulu, S.; Mostizky, Y.; Sunitha, S.; Shani, E.; Nahum, H.; Salomon, D.; Hayun, L.B.; Gruetter, C.; Rauh, D.; Ori, N.; et al. BSKs are partially redundant positive regulators of brassinosteroid signaling in Arabidopsis. Plant J. 2013, 74, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Ding, G.; Che, P.; Ilarslan, H.; Wurtele, E.S.; Nikolau, B.J. Genetic dissection of methylcrotonyl CoA carboxylase indicates a complex role for mitochondrial leucine catabolism during seed development and germination. Plant J. 2012, 70, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Pan, G.; Liu, H.; Wu, J.; Li, Y.; Zhao, Z.; Fu, T.; Zhou, Y. The chloroplast ribosomal protein L21 gene is essential for plastid development and embryogenesis in Arabidopsis. Planta 2012, 235, 907–921. [Google Scholar] [CrossRef]
- Hyten, D.L.; Pantalone, V.R.; Sams, C.E.; Saxton, A.M.; Landau-Ellis, D.; Stefaniak, T.R.; Schmidt, M.E. Seed quality QTL in a prominent soybean population. Theor. Appl. Genet. 2004, 109, 552–561. [Google Scholar] [CrossRef]
- Sun, Y.N.; Pan, J.B.; Shi, X.L.; Du, X.Y.; Wu, Q.; Qi, Z.M.; Jiang, H.W.; Xin, D.W.; Liu, C.Y.; Hu, G.H.; et al. Multi-environment mapping and meta-analysis of 100-seed weight in soybean. Mol. Biol. Rep. 2012, 39, 9435–9443. [Google Scholar] [CrossRef]
- Yan, L.; Li, Y.H.; Yang, C.Y.; Ren, S.X.; Chang, R.Z.; Zhang, M.C.; Qiu, L.J. Identification and validation of an over-dominant QTL controlling soybean seed weight using populations derived from Glycine max × Glycine soja. Plant Breed. 2014, 133, 632–637. [Google Scholar] [CrossRef]
- Maughan, P.J.; Saghai Maroof, M.A.; Buss, G.R. Molecular-marker analysis of seed-weight: Genomic locations, gene action, and evidence for orthologous evolution among three legume species. Theor. Appl. Genet. 1996, 93, 574–579. [Google Scholar] [CrossRef]
- Chen, Q.S.; Zhang, Z.C.; Liu, C.Y.; Xin, D.W.; Qiu, H.M.; Shan, D.P.; Shan, C.Y.; Hu, G.H. QTL analysis of major agronomic traits in soybean. Agric. Sci. China 2007, 6, 399–405. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, J.; Chen, S.; Shu, L.; Palmer, R.G.; Xing, G.; Li, Y.; Yang, S.; Yu, D.; Zhao, T.; et al. Exploration of presence/absence variation and corresponding polymorphic markers in soybean genome. J. Integr. Plant Biol. 2014, 56, 1009–1019. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Sayama, T.; Fujii, K.; Yumoto, S.; Kono, Y.; Hwang, T.Y.; Kikuchi, A.; Takada, Y.; Tanaka, Y.; Shiraiwa, T.; et al. A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds. Theor. Appl. Genet. 2014, 127, 1365–1374. [Google Scholar] [CrossRef]
- Li, D.; Sun, M.; Han, Y.; Teng, W.; Li, W. Identification of QTL underlying soluble pigment content in soybean stems related to resistance to soybean white mold (Sclerotinia sclerotiorum). Euphytica 2010, 172, 49–57. [Google Scholar] [CrossRef]
- Yao, D.; Liu, Z.Z.; Zhang, J.; Liu, S.Y.; Qu, J.; Guan, S.Y.; Pan, L.D.; Wang, D.; Liu, J.W.; Wang, P.W. Analysis of quantitative trait loci for main plant traits in soybean. Genet. Mol. Res. 2015, 14, 6101–6109. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Park, K.Y.; Lee, H.S.; Park, E.H.; Boerma, H.R. Genetic mapping of QTLs conditioning soybean sprout yield and quality. Theor. Appl. Genet. 2001, 103, 702–709. [Google Scholar] [CrossRef]
- Specht, J.E.; Chase, K.; Macrander, M.; Graef, G.L.; Chung, J.; Markwell, J.P.; Germann, M.; Orf, J.H.; Lark, K.G. Soybean response to water: A QTL analysis of drought tolerance. Crop Sci. 2001, 41, 493–509. [Google Scholar] [CrossRef]
- Li, W.; Zheng, D.; Van, K.; Lee, S. QTL mapping for major agronomic traits across two years in soybean (Glycine max L. Merr.). J. Crop Sci. Biotech. 2008, 11, 171–190. [Google Scholar]
- Pathan, S.M.; Vuong, T.; Clark, K.; Lee, J.D.; Grover Shannon, J.; Roberts, C.A.; Ellersieck, M.R.; Burton, J.W.; Cregan, P.B.; Hyten, D.L.; et al. Genetic mapping and confirmation of quantitative trait loci for seed protein and oil contents and seed weight in soybean. Crop Sci. 2013, 53, 765–774. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kaga, A.; Tomooka, N.; Yano, H.; Takada, Y.; Kato, S.; Vaughan, D. QTL affecting fitness of hybrids between wild and cultivated soybeans in experimental fields. Ecol. Evol. 2013, 3, 2150–2168. [Google Scholar] [CrossRef]
- Moongkanna, J.; Nakasathien, S.; Novitzky, W.P.; Kwanyuen, P.; Sinchaisri, P.; Srinives, P. SSR markers linking to seed traits and total oil content in soybean. Thai J. Agric. Sci. 2011, 44, 233–241. [Google Scholar]
- Funatsuki, H.; Kawaguchi, K.; Matsuba, S.; Sato, Y.; Ishimoto, M. Mapping of QTL associated with chilling tolerance during reproductive growth in soybean. Theor. Appl. Genet. 2005, 111, 851–861. [Google Scholar] [CrossRef]
- Orf, J.H.; Chase, K.; Jarvik, T.; Mansur, L.M.; Cregan, P.B.; Adler, F.R.; Lark, K.G. Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci. 1999, 39, 1642–1651. [Google Scholar] [CrossRef]
- Nichols, D.M.; Glover, K.D.; Carlson, S.R.; Specht, J.E.; Diers, B.W. Fine mapping of a seed protein QTL on soybean linkage group I and its correlated effects on agronomic traits. Crop Sci. 2006, 46, 834–839. [Google Scholar] [CrossRef]
- Kastoori, R.R.; Jedlicka, J.; Graef, G.L.; Waters, B.M. Identification of new QTLs for seed mineral, cysteine, and methionine concentrations in soybean [Glycine max (L.) Merr.]. Mol. Breed. 2014, 34, 431–445. [Google Scholar] [CrossRef]
- Xu, Y.; Yang, T.; Zhou, Y.; Yin, S.; Li, P.; Liu, J.; Xu, S.; Yang, Z.; Xu, C. Genome-wide association mapping of starch pasting properties in maize using single-locus and multi-locus models. Front. Plant Sci. 2018, 9, 1311. [Google Scholar] [CrossRef]
- Wang, J.; Wan, X.; Crossa, J.; Crouch, J.; Weng, J.; Zhai, H.; Wan, J. QTL mapping of grain length in rice (Oryza sativa L.) using chromosome segment substitution lines. Genet. Res. 2006, 88, 93–104. [Google Scholar] [CrossRef]
- Clouse, S.D. Brassinosteroid signal transduction: From receptor kinase activation to transcriptional networks regulating plant development. Plant Cell 2011, 23, 1219–1230. [Google Scholar] [CrossRef]
- Ohnishi, T.; Szatmari, A.M.; Watanabe, B.; Fujita, S.; Bancos, S.; Koncz, C.; Lafos, M.; Shibata, K.; Yokota, T.; Sakata, K.; et al. C-23 hydroxylation by Arabidopsis CYP90C1 and CYP90D1 reveals a novel shortcut in brassinosteroid biosynthesis. Plant Cell 2006, 18, 3275–3288. [Google Scholar] [CrossRef]
- Yang, C.J.; Zhang, C.; Lu, Y.N.; Jin, J.Q.; Wang, X.L. The mechanisms of brassinosteroids’ action: From signal transduction to plant development. Mol. Plant 2011, 4, 588–600. [Google Scholar] [CrossRef]
- Hirshfield, K.; Flannery, R.; Daie, J. Cotyledon cell number and cell size in relation to seed size and seed yield of soybean. Plant Physiol. Biochem. 1993, 31, 395–400. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Environment | Mean | Min | Max | SD | CV (%) | Skew | Kur | FG | FE | h2B (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| E1, Nanjing 2014 | 18.39 | 6.18 | 44.53 | 5.96 | 32.39 | 0.95 | 1.07 | 25.43 ** | 18.65 ** | 93.70 |
| E2, Nanjing 2015 | 19.86 | 5.28 | 40.64 | 5.58 | 28.08 | 0.49 | 0.62 | 88.51 | ||
| E3, Wuhan 2014 | 17.98 | 6.75 | 37.57 | 5.07 | 28.20 | 0.69 | 1.09 | 90.15 | ||
| E4, Wuhan 2015 | 19.22 | 8.83 | 51.19 | 5.58 | 29.07 | 1.03 | 1.13 | 83.23 |
| Method | E1 | E2 | E3 | E4 | BLUP | QTN Effect | LOD Score | r2 (%) |
|---|---|---|---|---|---|---|---|---|
| Total | 66 | 76 | 45 | 55 | 70 | −4.66–4.07 | 3.01–18.08 | 0.38–7.88 |
| mrMLM | 20 | 19 | 15 | 16 | 13 | −4.66–2.11 | 3.01–11.72 | 0.72–6.94 |
| FASTmrMLM | 18 | 22 | 14 | 12 | 24 | −2.60–2.31 | 3.01–13.80 | 0.49–7.88 |
| FASTmrEMMA | 10 | 10 | 9 | 8 | 13 | −3.48–4.07 | 3.18–12.96 | 1.02–7.26 |
| pLARmEB | 21 | 18 | 16 | 18 | 20 | −3.98–2.54 | 3.03–15.34 | 0.38–6.90 |
| pKWmEB | 17 | 25 | 17 | 18 | 20 | −2.55–1.88 | 3.04–12.84 | 1.04–5.52 |
| ISIS EM-BLASSO | 22 | 18 | 11 | 16 | 22 | −4.61–1.73 | 3.11–18.08 | 0.66–5.91 |
| QTN a | Position (bp) | Effect | LOD Score | r2 (%) b | MAF | Method c | Environment d | SW Increasing Allele | Average 100-Seed Weight | SSR Marker | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SW Increasing Allele | SW Decreasing Allele | Total | Significance e | ||||||||||
| qcHSW-1-1 | Gm01_6389301-6594737 | 1.36–4.07 | 4.71–6.30 | 2.93–4.94 | 0.09 | 1–3, 5, 6 | E1–E3, BLUP | A | 18.34–20.29 | 12.05–13.95 | 17.98–19.86 | ** | BARCSOYSSR_01_0337 |
| qHSW-2-2 | Gm02_43506875 | 0.83–2.77 | 3.14–5.66 | 1.06–2.53 | 0.13 | 1–6 | E1, E3, E4, BLUP | C | 23.23–24.20 | 17.91–19.56 | 18.3919.86 | ** | BARCSOYSSR_02_1373 |
| qHSW-3-1 | Gm03_17127049 | 0.76–1.49 | 3.86–6.09 | 0.61–1.67 | 0.1 | 2, 4 | E1, E2, BLUP | G | 22.50–24.89 | 17.45–17.68 | 17.98–18.39 | ** | BARCSOYSSR_03_0519 |
| qHSW-3-2 | Gm03_29644613 | 0.54–0.82 | 3.67–6.65 | 1.49–2.44 | 0.33 | 2, 5, 6 | E1, E2, BLUP | T | 19.12–19.48 | 16.05–16.51 | 17.98–18.39 | ** | BARCSOYSSR_03_0762 |
| qHSW-3-3 | Gm03_37435877 | −2.55–1.35 | 3.18–4.98 | 0.67–3.24 | 0.02 | 2, 5 | E1, E4, BLUP | C | 26.24–27.94 | 18.20–19.01 | 18.39–19.22 | ** | BARCSOYSSR_03_1155 |
| qHSW-4-1 | Gm04_6703334 | −3.33–0.48 | 3.01–8.64 | 1.05–5.34 | 0.2 | 1–6 | E1–E4, BLUP | C | 21.37–24.09 | 17.20–18.93 | 17.98–19.22 | ** | BARC-025825-05102 |
| qHSW-4-2 | Gm04_37026887 | 0.59–1.71 | 3.34–9.93 | 1.11–4.20 | 0.26 | 1–3 | E1–E3, BLUP | A | 19.29–21.24 | 14.54–16.33 | 17.98–19.86 | ** | BARCSOYSSR_04_0940 |
| qHSW-4-3 | Gm04_39207103 | −1.19–0.75 | 4.09–6.23 | 1.22–4.75 | 0.15 | 1, 2, 4, 5 | E1, E2, E4 | C | 21.50–22.39 | 17.36–18.61 | 17.98–19.22 | ** | BARCSOYSSR_04_1006 |
| qHSW-5-1 | Gm05_38490635 | 0.62–1.24 | 3.29–8.13 | 1.24–2.94 | 0.47 | 2, 3 | E1, E3, BLUP | A | 19.64–20.73 | 17.00–18.85 | 18.39–19.86 | * | BARCSOYSSR_05_1241 |
| qHSW-5-2 | Gm05_40410433 | 0.68–1.20 | 5.24–6.88 | 1.35–3.4 | 0.22 | 6 | E1, E4, BLUP | A | 19.44–20.16 | 14.29–15.53 | 18.39–19.22 | ** | BARC-007964-00162 |
| qHSW-6-1 | Gm06_5910218 | 0.71–2.71 | 3.50–11.68 | 1.61–6.94 | 0.4 | 1–6 | E1–E3, BLUP | G | 19.38–21.49 | 15.71–17.24 | 17.39–19.86 | ** | BARC-045145-08894 |
| qcHSW-6-3 | Gm06_13385023-13385940 | 0.813–3.44 | 3.13–6.90 | 0.97–3.59 | 0.13 | 2–6 | E1–E3, BLUP | T | 18.53–20.49 | 13.87–15.87 | 18.39–19.86 | ** | BARCSOYSSR_06_0614 |
| qHSW-6-4 | Gm06_14086552 | 0.45–2.37 | 3.03–9.43 | 0.63–5.13 | 0.38 | 1–6 | E1, E2, BLUP | C | 19.16–19.99 | 15.81–16.07 | 17.98–18.39 | ** | BARCSOYSSR_06_0759 |
| qcHSW-7-3 | Gm07_6439407-6463021 | 0.94–2.25 | 4.56–18.08 | 2.42–5.91 | 0.41 | 1–6 | E1–E4, BLUP | T | 19.74–21.58 | 16.79–18.74 | 17.98–18.39 | ** | BARCSOYSSR_07_0334 |
| qHSW-8-1 | Gm08_10314889 | 0.46–0.76 | 3.17–4.33 | 0.68–1.79 | 0.28 | 1, 5, 6 | E1, E2, E4, BLUP | C | 19.97–21.47 | 17.27–18.40 | 17.98–19.22 | ** | BARC-038631-07266 |
| qcHSW-10-4 | Gm10_44349893-44469282 | 0.66–2.34 | 3.26–10.73 | 1.37–5.90 | 0.27 | 1–6 | E1–E4, BLUP | C | 20.08–21.88 | 17.23–19.28 | 17.98–19.22 | ** | BARCSOYSSR_10_1419 |
| qHSW-11-1 | Gm11_5245829 | 0.50–0.77 | 3.38–7.34 | 1.20–2.27 | 0.39 | 5, 6 | E1, E2, BLUP | G | 19.67–20.24 | 15.62–15.76 | 17.98–18.39 | ** | BARC-018099-02516 |
| qHSW-11-2 | Gm11_9337224 | 0.55–2.95 | 3.09–11.03 | 1.04–5.50 | 0.32 | 1–6 | E1, E2, BLUP | A | 20.10–21.16 | 16.95–17.06 | 17.98–18.39 | ** | BARCSOYSSR_11_0511 |
| qHSW-11-3 | Gm11_11100801 | −1.39–0.75 | 3.19–5.25 | 1.34–2.88 | 0.12 | 5 | E1, E2, E4, BLUP | A | 22.35–24.23 | 17.38–18.54 | 17.98–19.22 | ** | BARCSOYSSR_11_0615 |
| qHSW-14-1 | Gm14_10032197 | 0.79–1.52 | 3.77–15.34 | 1.36–6.90 | 0.25 | 4 | E1, E2, BLUP | C | 19.13–19.75 | 14.32–14.54 | 17.98–18.39 | ** | BARC-052759-11611 |
| qHSW-17-6 | Gm17_38985524 | −3.38–1.16 | 4.36–9.49 | 1.79–4.60 | 0.1 | 1–6 | E3, E4, BLUP | A | 23.35–23.90 | 18.76–18.77 | 19.22–19.86 | ** | BARCSOYSSR_17_1474 |
| qHSW-20-2 | Gm20_45498156 | 0.76–1.82 | 3.26–10.33 | 1.93–5.09 | 0.37 | 1–3, 5 | E1, E2, BLUP | G | 19.20–20.07 | 16.33–16.63 | 17.98–18.39 | ** | BARC-047899-10425 |
| Breeding Objective | Predicted Parental Combinations |
|---|---|
| Large seed | Yafanzaodou × Ribendaheidou |
| Nannong 95C-5 × Ribendaheidou | |
| Quxiandahuangdou × Yixingwuhuangdou | |
| Bayueqing × Fujiandadou | |
| Nanchengqingpidadou × Ribendaheidou | |
| Small seed | Heibiqing × Mayidan |
| Qinyan 1 × Mayidan | |
| Qingcha 1× Mayidan | |
| Qinyan 1 × Heibiqing | |
| Mingshanhongxingjiroudou × Mayidan |
| Genome-Wide Association Study | Soybean Genes | Comparative Genomic Study | KEGG Pathway | Reference | ||||
|---|---|---|---|---|---|---|---|---|
| QTN (QTN Cluster) | Position (bp) | Candidate Gene | Position (bp) | Gene Name | Arabidopsis Gene | Functional Annotation | ||
| qHSW-5-1 | Gm05_38490635 | Glyma05g34120 | Gm05:38540979-38549756 | Translation elongation factor EF1A | mRNA surveillance pathway | [7] | ||
| qHSW-6-2 | Gm06_8258824 | Glyma06g10830 | Gm06:8199290-8204935 | GmCRY1 | cryptochrome 1 | Circadian rhythm—plant | [37] | |
| qcHSW-6-3 | Gm06_13385023-13385940 | Glyma06g16920 | Gm06:13300048-13304817 | GmCPK11 | calcium-dependent protein kinase 2 | Plant-pathogen interaction | [66] | |
| qcHSW-7-3 | Gm07_6439407-6463021 | Glyma07g07850 | Gm07:6497229-6504722 | BSK3 | AT4G00710 | BR-signaling kinase 3 | Plant hormone signal transduction | [67] |
| qcHSW-10-1 | Gm10_2563422-2566365 | Glyma10g03440 | Gm10:2482369-2489108 | MCCA | AT1G03090 | methylcrotonyl-CoA carboxylase alpha chain | Metabolic pathways | [68] |
| qcHSW-10-4 | Gm10_44349893-44469282 | Glyma10g36070 | Gm10:44258213-44261525 | RPL21 | AT1G35680 | Ribosomal protein L21 | Ribosome | [69] |
| QTN or QTN Cluster in This Study | Previously Reported QTLs | QTN or QTN Cluster in This Study | Previously Reported QTLs | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Name | Position (bp) | Name | Marker Associated | Reference | Name | Position (bp) | Name | Marker Associated | Reference |
| qcHSW-1-1 | Gm01_6389301-6594737 | SW 15-2 | Sat_305-Satt531 | [70] | qcHSW-10-4 | Gm10_44349893-44469282 | SW 34-8 | BARC-028651-05984-Satt479 | [14] |
| qHSW-2-1 | Gm02_11278769 | SW 49-8 | Satt172-Satt157 | [17] | qHSW-11-1 | Gm11_5245829 | SW 37-9 | Sat_149-BARC-029533-06211 | [71] |
| qHSW-2-2 | Gm02_43506875 | SW 49-8 | Satt172-Satt157 | [17] | qHSW-11-2 | Gm11_9337224 | SW 37-9 | Sat_149-BARC-029533-06211 | [71] |
| qHSW-4-2 | Gm04_37026887 | SW 45-3 | Sat_042-Sat_322 | [72] | qHSW-11-3 | Gm11_11100801 | SW 11-1 | BARC-059851-16137-BARC-016279-02316 | [79] |
| qHSW-4-3 | Gm04_39207103 | SW 45-3 | Sat_042-Sat_322 | [72] | qHSW-11-4 | Gm11_27803417 | SW 10-3 | Satt415-BARC-041167-07925 | [80] |
| qHSW-5-1 | Gm05_38490635 | SW 10-1 | BARC-060051-16321-BARC-045267-08918 | [80] | qHSW-14-1 | Gm14_10032197 | SW 23-1 | Satt601- BARC-059265-15700 | [81] |
| qHSW-5-2 | Gm05_40410433 | SW 36-10 | BARC-029873-06450-BARC-027778-06658 | [14] | qHSW-17-1 | Gm17_8760885 | SW 42-2 | Satt154- BARC-058841-15463 | [75] |
| qHSW-6-1 | Gm06_5910218 | cqSW-008 | Satt457-BARC-059997-16280 | [82] | qHSW-17-2 | Gm17_12908030 | SW 43-2 | Satt389-Satt447 | [83] |
| qHSW-6-2 | Gm06_8258824 | SW 33-1 | Sat_153-Satt291 | [84] | qHSW-17-3 | Gm17_13325606 | SW 43-2 | Satt389-Satt447 | [83] |
| qcHSW-6-3 | Gm06_13385023-13385940 | SW 4-1 | Sat_246-Satt640 | [73] | qHSW-17-4 | Gm17_16770188 | SW 47-2 | Satt256-Satt458 | [77] |
| qHSW-6-4 | Gm06_14086552 | SW 16-1 | Sat_238-BARC-014491-01561 | [85] | qHSW-17-5 | Gm17_38229512 | SW 49-10 | BARC-013709-01242-BARC-056481-14397 | [17] |
| qHSW-7-1 | Gm07_2535953 | SW 7-6 | Sat_316-Satt201 | [86] | qHSW-17-6 | Gm17_38985524 | SW 49-10 | BARC-013709-01242-BARC-056481-14397 | [17] |
| qHSW-7-2 | Gm07_3954121 | SW 49-15 | Satt323-Satt150 | [17] | qHSW-18-1 | Gm18_5591131 | SW 50-4 | Satt115-Sat_315 | [76] |
| qcHSW-7-3 | Gm07_6439407-6463021 | SW 45-4 | BARC-039383-07310-Satt567 | [72] | qHSW-18-2 | Gm18_6705051 | SW/p 6-5 | Sat_308-Satt324 | [78] |
| qHSW-8-1 | Gm08_10314889 | SW 34-10 | Satt424-Satt390 | [14] | qHSW-20-1 | Gm20_30017454 | cqSW-003 | BARC-041129-07912-Satt127 | [87] |
| qcHSW-10-2 | Gm10_32393792-32584066 | SW 53-1 | BARC-064941-19017-BARC-051153-11022 | [88] | qHSW-20-2 | Gm20_45498156 | SW 50-16 | Sct_189-Satt623 | [76] |
| qHSW-10-3 | Gm10_42750933 | SW 25-4 | BARC-037165-06725-Satt173 | [74] | |||||
| Group | No. of QTNs | Absolute Effect | LOD Score | r2 (%) | % Known QTNs |
|---|---|---|---|---|---|
| 1 (Both esQTN and msQTN) | 15 | 1.56 ± 1.10 | 6.29 ± 3.65 | 2.99 ± 1.94 | 86.67 |
| 2 (esQTN rather than msQTN) | 7 | 1.12 ± 0.54 | 5.70 ± 3.22 | 2.21 ± 1.63 | 71.42 |
| 3 (msQTN rather than esQTN) | 21 | 1.35 ± 0.87 | 6.38 ± 3.10 | 2.86 ± 1.71 | 66.67 |
| 4 (Neither esQTN nor msQTN) | 14 | 1.28 ± 1.01 | 5.43 ± 2.16 | 2.35 ± 1.46 | 50.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikram, M.; Han, X.; Zuo, J.-F.; Song, J.; Han, C.-Y.; Zhang, Y.-W.; Zhang, Y.-M. Identification of QTNs and Their Candidate Genes for 100-Seed Weight in Soybean (Glycine max L.) Using Multi-Locus Genome-Wide Association Studies. Genes 2020, 11, 714. https://doi.org/10.3390/genes11070714
Ikram M, Han X, Zuo J-F, Song J, Han C-Y, Zhang Y-W, Zhang Y-M. Identification of QTNs and Their Candidate Genes for 100-Seed Weight in Soybean (Glycine max L.) Using Multi-Locus Genome-Wide Association Studies. Genes. 2020; 11(7):714. https://doi.org/10.3390/genes11070714
Chicago/Turabian StyleIkram, Muhammad, Xu Han, Jian-Fang Zuo, Jian Song, Chun-Yu Han, Ya-Wen Zhang, and Yuan-Ming Zhang. 2020. "Identification of QTNs and Their Candidate Genes for 100-Seed Weight in Soybean (Glycine max L.) Using Multi-Locus Genome-Wide Association Studies" Genes 11, no. 7: 714. https://doi.org/10.3390/genes11070714
APA StyleIkram, M., Han, X., Zuo, J.-F., Song, J., Han, C.-Y., Zhang, Y.-W., & Zhang, Y.-M. (2020). Identification of QTNs and Their Candidate Genes for 100-Seed Weight in Soybean (Glycine max L.) Using Multi-Locus Genome-Wide Association Studies. Genes, 11(7), 714. https://doi.org/10.3390/genes11070714