Array-Based Epigenetic Aging Indices May Be Racially Biased


 ,
,
Abstract
1. Introduction
2. Materials and Methods
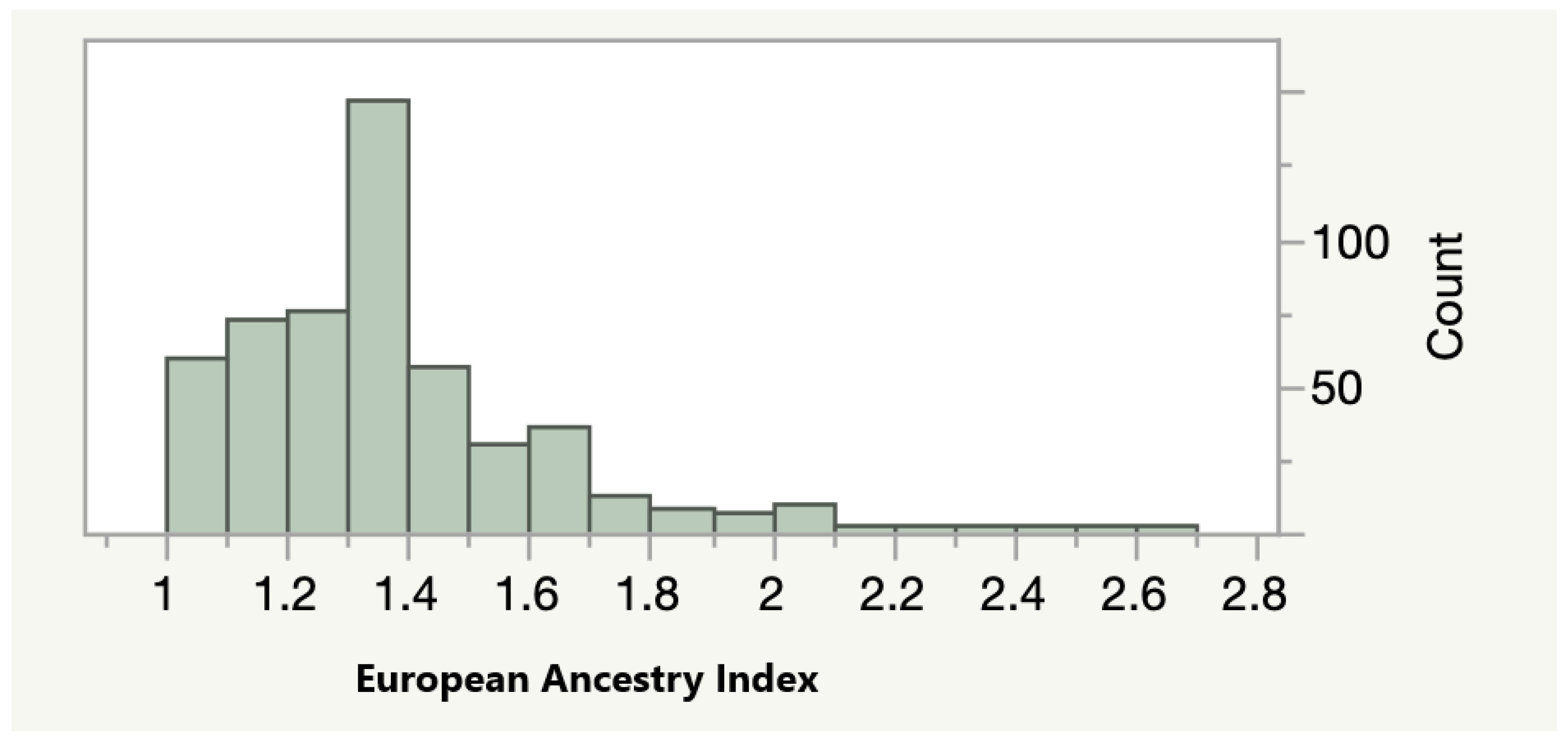
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dedeurwaerder, S.; Defrance, M.; Bizet, M.; Calonne, E.; Bontempi, G.; Fuks, F. A comprehensive overview of Infinium HumanMethylation450 data processing. Brief. Bioinform. 2014, 15, 929–941. [Google Scholar] [CrossRef]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef]
- Soriano-Tárraga, C.; Jiménez-Conde, J.; Giralt-Steinhauer, E.; Mola-Caminal, M.; Vivanco-Hidalgo, R.M.; Ois, A.; Rodríguez-Campello, A.; Cuadrado-Godia, E.; Sayols-Baixeras, S.; Elosua, R. Epigenome-wide association study identifies TXNIP gene associated with type 2 diabetes mellitus and sustained hyperglycemia. Hum. Mol. Genet. 2016, 25, 609–619. [Google Scholar] [CrossRef]
- Monick, M.M.; Beach, S.R.; Plume, J.; Sears, R.; Gerrard, M.; Brody, G.H.; Philibert, R.A. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am. J. Med. Genet. 2012, 159, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Subirana, I.; Degano, I.R.; Elosua, R. Association between DNA methylation and coronary heart disease or other atherosclerotic events: A systematic review. Atherosclerosis 2017, 263, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Paul, K.C.; Chuang, Y.-H.; Cockburn, M.; Bronstein, J.M.; Horvath, S.; Ritz, B. Organophosphate pesticide exposure and differential genome-wide DNA methylation. Sci. Total Environ. 2018, 645, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Marioni, R.E.; Hedman, Å.K.; Pfeiffer, L.; Tsai, P.-C.; Reynolds, L.M.; Just, A.C.; Duan, Q.; Boer, C.G.; Tanaka, T. A DNA methylation biomarker of alcohol consumption. Mol. Psychiatry 2018, 23, 422–433. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Philibert, R.; Penaluna, B.; White, T.; Shires, S.; Gunter, T.D.; Liesveld, J.; Erwin, C.; Hollenbeck, N.; Osborn, T. A pilot examination of the genome-wide DNA methylation signatures of subjects entering and exiting short-term alcohol dependence treatment programs. Epigenetics 2014, 9, 1212–1219. [Google Scholar] [CrossRef]
- Bock, C.; Halbritter, F.; Carmona, F.J.; Tierling, S.; Datlinger, P.; Assenov, Y.; Berdasco, M.; Bergmann, A.K.; Booher, K.; Busato, F. Quantitative comparison of DNA methylation assays for biomarker development and clinical applications. Nat. Biotechnol. 2016, 34, 726. [Google Scholar]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Esteller, M. Epigenetics and aging: The targets and the marks. Trends Genet. 2007, 23, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 2018, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological age predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371. [Google Scholar] [CrossRef]
- Philibert, R.A.; Terry, N.; Erwin, C.; Philibert, W.J.; Beach, S.R.; Brody, G.H. Methylation array data can simultaneously identify individuals and convey protected health information: An unrecognized ethical concern. Clin. Epigenetics 2014, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, E.; Shenhav, L.; Schweiger, R.; Yousefi, P.; Huen, K.; Eskenazi, B.; Eng, C.; Huntsman, S.; Hu, D.; Galanter, J.; et al. Genome-wide methylation data mirror ancestry information. Epigenetics Chromatin 2017, 10, 1. [Google Scholar] [CrossRef]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Infinium HumanMethylation450K v1.2 Product Files. Available online: https://support.illumina.com/downloads/infinium_humanmethylation450_product_files.html (accessed on 26 April 2020).
- Czamara, D.; Eraslan, G.; Page, C.M.; Lahti, J.; Lahti-Pulkkinen, M.; Hämäläinen, E.; Kajantie, E.; Laivuori, H.; Villa, P.M.; Reynolds, R.M. Integrated analysis of environmental and genetic influences on cord blood DNA methylation in new-borns. Nat. Commun. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Dogan, M.V.; Beach, S.R.; Philibert, R.A. Genetically contextual effects of smoking on genome wide DNA methylation. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2017, 174, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Xiang, J.; Beach, S.R.; Cutrona, C.; Gibbons, F.X.; Simons, R.L.; Brody, G.H.; Stapleton, J.T.; Philibert, R.A. Ethnicity and smoking-associated DNA methylation changes at HIV co-receptor GPR15. Front. Psychiatry 2015, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.A.; Beach, S.R.; Dogan, M.; Simons, R.L.; Gibbons, F.X.; Long, J.D.; Philibert, R. A direct comparison of the relationship of epigenetic aging and epigenetic substance consumption markers to mortality in the Framingham heart study. Genes 2019, 10, 51. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.-K.; Gibbons, F.X.; Simons, R.L.; Philibert, R.A.; Beach, S.R. The Effect of Tobacco Smoking Differs across Indices of DNA Methylation-Based Aging in an African American Sample: DNA Methylation-Based Indices of Smoking Capture These Effects. Genes 2020, 11, 311. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 2019, 11, 303. [Google Scholar] [CrossRef]
- Horsthemke, B. A critical view on transgenerational epigenetic inheritance in humans. Nat. Commun. 2018, 9, 1–4. [Google Scholar] [CrossRef]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef]
- Mozhui, K.; Smith, A.K.; Tylavsky, F.A. Ancestry dependent DNA methylation and influence of maternal nutrition. PLoS ONE 2015, 10, e0118466. [Google Scholar] [CrossRef]
- Simons, R.L.; Lei, M.K.; Beach, S.R.; Brody, G.H.; Philibert, R.A.; Gibbons, F.X. Social environmental variation, plasticity genes, and aggression: Evidence for the differential susceptibility hypothesis. Am. Sociol. Rev. 2011, 76, 833. [Google Scholar] [CrossRef]
- Gibbons, F.X.; Gerrard, M.; Cleveland, M.J.; Wills, T.A.; Brody, G. Perceived discrimination and substance use in African American parents and their children: A panel study. J. Pers. Soc. Psychol. 2004, 86, 517. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Nassir, R.; Kosoy, R.; Tian, C.; White, P.A.; Butler, L.M.; Silva, G.; Kittles, R.; Alarcon-Riquelme, M.E.; Gregersen, P.K.; Belmont, J.W. An ancestry informative marker set for determining continental origin: Validation and extension using human genome diversity panels. BMC Genet. 2009, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.A.; McNeil, B.J. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology 1983, 148, 839–843. [Google Scholar] [CrossRef]
- Fleiss, J.L. Statistical Methods for Rates and Proportions, 2nd ed.; John Wiley & Sons Inc: New York, NY, USA, 1981. [Google Scholar]
- Huan, T.; Joehanes, R.; Song, C.; Peng, F.; Guo, Y.; Mendelson, M.; Yao, C.; Liu, C.; Ma, J.; Richard, M. Genome-wide identification of DNA methylation QTLs in whole blood highlights pathways for cardiovascular disease. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Prevention: Vital signs: Current cigarette smoking among adults aged ≥ 18 years--United States, 2005–2010. MMWR Morb. Mortal. Wkly. Rep. 2011, 60, 1207. [Google Scholar]
- Grucza, R.A.; Sher, K.J.; Kerr, W.C.; Krauss, M.J.; Lui, C.K.; McDowell, Y.E.; Hartz, S.; Virdi, G.; Bierut, L.J. Trends in adult alcohol use and binge drinking in the early 21st-century United States: A meta-analysis of 6 National Survey Series. Alcohol. Clin. Exp. Res. 2018, 42, 1939–1950. [Google Scholar] [CrossRef]
- Bryc, K.; Durand, E.Y.; Macpherson, J.M.; Reich, D.; Mountain, J.L. The genetic ancestry of african americans, latinos, and european Americans across the United States. Am. J. Hum. Genet. 2015, 96, 37–53. [Google Scholar] [CrossRef] [PubMed]
- Dogan, M.V.; Shields, B.; Cutrona, C.; Gao, L.; Gibbons, F.X.; Simons, R.; Monick, M.; Brody, G.; Tan, K.; Philibert, R. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genom. 2014, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Schalkwyk, L.C.; Meaburn, E.L.; Smith, R.; Dempster, E.L.; Jeffries, A.R.; Davies, M.N.; Plomin, R.; Mill, J. Allelic skewing of DNA methylation is widespread across the genome. Am. J. Hum. Genet. 2010, 86, 196–212. [Google Scholar] [CrossRef]
- Infinium HumanMethylation450K BeadChip Product Files. Available online: https://support.illumina.com/array/array_kits/infinium_humanmethylation450_beadchip_kit/downloads.html (accessed on 26 April 2020).
- Yuan, V.; Price, E.M.; Del Gobbo, G.; Mostafavi, S.; Cox, B.; Binder, A.M.; Michels, K.B.; Marsit, C.; Robinson, W.P. Accurate ethnicity prediction from placental DNA methylation data. Epigenetics Chromatin 2019, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lövkvist, C.; Dodd, I.B.; Sneppen, K.; Haerter, J.O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016, 44, 5123–5132. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Tang, B.; Tang, Y. Allele-specific genome targeting in the development of precision medicine. Theranostics 2020, 10, 3118. [Google Scholar] [CrossRef]
- Zhang, Y.; Saum, K.-U.; Schöttker, B.; Holleczek, B.; Brenner, H. Methylomic survival predictors, frailty, and mortality. Aging (Albany NY) 2018, 10, 339. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Miller, S.; Noel, A.; Dawes, K.; Papworth, E.; Black, D.W.; Beach, S.R.; Long, J.D.; Mills, J.A.; Dogan, M. A Four Marker Digital PCR Toolkit for Detecting Heavy Alcohol Consumption and the Effectiveness of Its Treatment. J. Insur. Med. 2019, 48, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Philibert, R.; Dogan, M.; Beach, S.R.; Mills, J.A.; Long, J.D. AHRR methylation predicts smoking status and smoking intensity in both saliva and blood DNA. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2020, 183, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Dupras, C.; Beck, S.; Rothstein, M.A.; Berner, A.; Saulnier, K.M.; Pinkesz, M.; Prince, A.E.; Liosi, S.; Song, L.; Joly, Y. Potential (mis) use of epigenetic age estimators by private companies and public agencies: Human rights law should provide ethical guidance. Environ. Epigenetics 2019, 5, dvz018. [Google Scholar]


{kind=link}
| Illumina Probe ID | t-Tests | BF Corrected | CHR | SNPs within 50 bp * | SNPs within 10 bp ** |
|---|---|---|---|---|---|
| cg08654655 | 2.28 × 10−15 | 1.17 × 10−12 | 1 | ||
| cg18771300 | 3.13 × 10−15 | 1.61 × 10−12 | 14 | ||
| cg15344028 | 9.86 × 10−15 | 5.06 × 10−12 | 2 | ||
| cg02016419 | 2.05 × 10−12 | 1.05 × 10−9 | 17 | ||
| cg12402251 | 4.56 × 10−12 | 2.35 × 10−9 | 8 | ||
| cg04718414 | 5.78 × 10−12 | 2.96 × 10−9 | 13 | rs17337675 | |
| cg08251399 | 1.81 × 10−11 | 9.29 × 10−9 | 2 | ||
| cg12864235 | 2.6 × 10−11 | 1.34 × 10−8 | 5 | ||
| cg09799873 | 2.26 × 10−10 | 1.16 × 10−7 | 19 | rs73925316 | |
| cg00862290 | 4.37 × 10−10 | 2.24 × 10−7 | 3 | ||
| cg06638451 | 5.77 × 10−10 | 2.96 × 10−7 | 3 | rs17059410 | |
| cg19566405 | 7.70 × 10−10 | 3.95 × 10−7 | 17 | ||
| cg13509147 | 7.77 × 10−10 | 3.99 × 10−7 | 19 | ||
| cg20066677 | 1.10 × 10−9 | 5.62 × 10−7 | 12 | ||
| cg16713727 | 3.18 × 10−8 | 1.63 × 10−5 | 1 | ||
| cg11618577 | 3.49 × 10−8 | 1.79 × 10−5 | 2 | ||
| cg12813792 | 4.94 × 10−8 | 2.53 × 10−5 | 20 | ||
| cg04836038 | 7.23 × 10−8 | 3.71 × 10−5 | 13 | ||
| cg27187881 | 8.21 × 10−8 | 4.21 × 10−5 | 22 | ||
| cg10795646 | 9.29 × 10−8 | 4.77 × 10−5 | 1 | ||
| cg15201877 | 2.32 × 10−7 | 0.0001191 | 1 | ||
| cg17133388 | 4.10 × 10−7 | 0.0002104 | 3 | ||
| cg13119609 | 4.64 × 10−7 | 0.000238 | 19 | ||
| cg22736354 | 4.96 × 10−7 | 0.0002545 | 6 | rs28940575 | |
| cg23159337 | 8.61 × 10−7 | 0.0004416 | 3 | rs34959916 | |
| cg24125648 | 1.21 × 10−6 | 0.000618 | 15 | rs75056397 | |
| cg15963417 | 1.38 × 10−6 | 0.0007092 | 12 | rs62652660 | |
| cg09304040 | 1.58 × 10−6 | 0.0008097 | 12 | ||
| cg09404633 | 1.82 × 10−6 | 0.0009359 | 1 | ||
| cg10570177 | 2.56 × 10−6 | 0.0013111 | 9 | rs36223203 |
| ID | p-Value | Associated with Smoking in FHS a |
|---|---|---|
| cg06638451 | 0.0002941 | No |
| cg04718414 | 0.0004285 | Yes |
| cg00168942 | 0.0008096 | Yes |
| cg00862290 | 0.0035711 | No |
| cg10795646 | 0.0104504 | No |
| cg19514469 | 0.0115081 | Yes |
| cg08251399 | 0.0149561 | Yes |
| cg09404633 | 0.0241233 | No |
| cg08067365 | 0.0354048 | No |
| cg07038400 | 0.040688 | No |
| cg03991512 | 0.0426108 | Yes |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philibert, R.; Beach, S.R.H.; Lei, M.-K.; Gibbons, F.X.; Gerrard, M.; Simons, R.L.; Dogan, M.V. Array-Based Epigenetic Aging Indices May Be Racially Biased. Genes 2020, 11, 685. https://doi.org/10.3390/genes11060685
Philibert R, Beach SRH, Lei M-K, Gibbons FX, Gerrard M, Simons RL, Dogan MV. Array-Based Epigenetic Aging Indices May Be Racially Biased. Genes. 2020; 11(6):685. https://doi.org/10.3390/genes11060685
Chicago/Turabian StylePhilibert, Robert, Steven R.H. Beach, Man-Kit Lei, Frederick X. Gibbons, Meg Gerrard, Ronald L. Simons, and Meeshanthini V. Dogan. 2020. "Array-Based Epigenetic Aging Indices May Be Racially Biased" Genes 11, no. 6: 685. https://doi.org/10.3390/genes11060685
APA StylePhilibert, R., Beach, S. R. H., Lei, M.-K., Gibbons, F. X., Gerrard, M., Simons, R. L., & Dogan, M. V. (2020). Array-Based Epigenetic Aging Indices May Be Racially Biased. Genes, 11(6), 685. https://doi.org/10.3390/genes11060685