Direct Visualization of Horizontal Gene Transfer by Transformation in Live Pneumococcal Cells Using Microfluidics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Culture and Transformation Conditions
2.2. Strain Constructions
2.3. Time-Lapse Microfluidic Microscopy
2.4. Image Analysis
2.5. Direct Visualization of Transformation Assay
3. Results
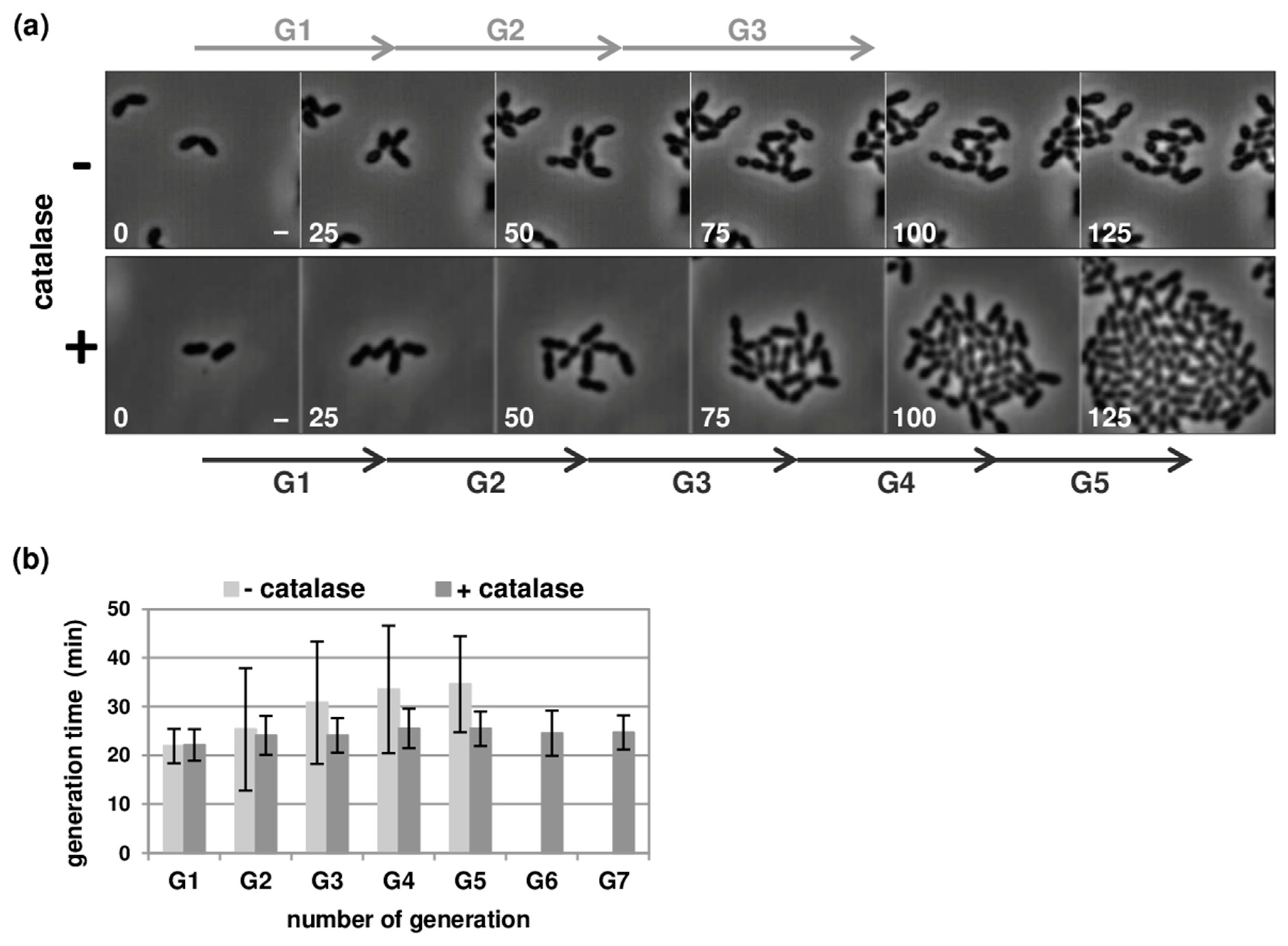
3.1. Optimisation of Pneumococcal Growth in Microfluidic Chambers
3.2. Robust Competence Development in Microfluidic Chambers
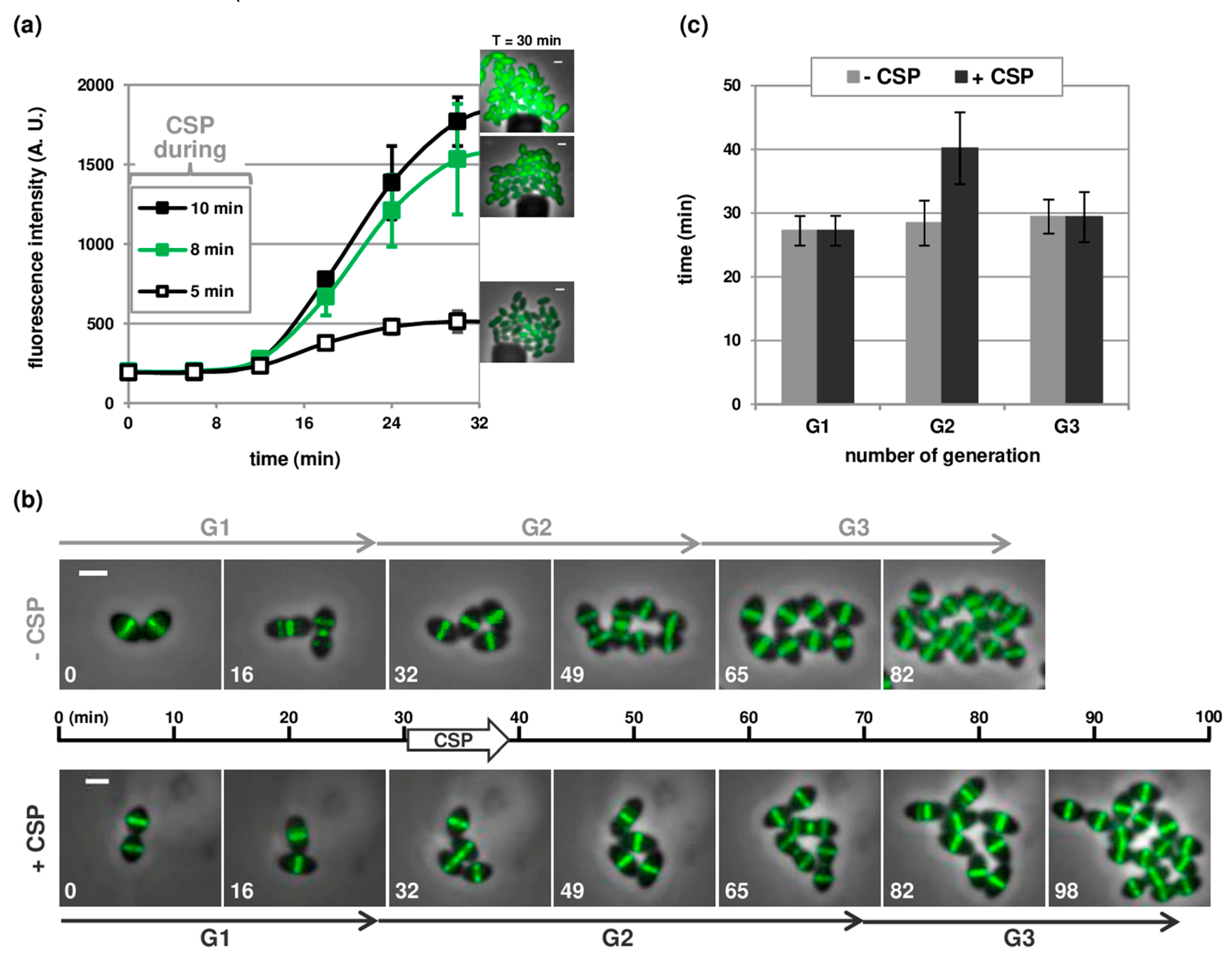
3.3. Temporal Visualization of Early and Late Com Genes Expression
3.4. Near Real-Time Visualization of Pneumococcal Transformation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnston, C.; Martin, B.; Fichant, G.; Polard, P.; Claverys, J.-P. Bacterial transformation: Distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014, 12, 181–196. [Google Scholar] [CrossRef]
- Dubnau, D.; Blokesch, M. Mechanisms of DNA Uptake by Naturally Competent Bacteria. Annu. Rev. Genet. 2019, 53, 217–237. [Google Scholar] [CrossRef]
- Griffith, F. The Significance of Pneumococcal Types. J. Hyg. 1928, 27, 113–159. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Finkelstein, J.A.; Pelton, S.I.; Mitchell, P.K.; Lee, G.M.; Parkhill, J.; Bentley, S.D.; Hanage, W.P.; Lipsitch, M. Population genomics of post-vaccine changes in pneumococcal epidemiology. Nat. Genet. 2013, 45, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Prudhomme, M.; Berge, M.; Martin, B.; Polard, P. Pneumococcal Competence Coordination Relies on a Cell-Contact Sensing Mechanism. PLOS Genet. 2016, 12, e1006113. [Google Scholar] [CrossRef]
- Håvarstein, L.S.; Coomaraswamy, G.; Morrison, D.A. An unmodified heptadecapeptide pheromone induces competence for genetic transformation in Streptococcus pneumoniae. Proc. Natl. Acad. Sci. USA 1995, 92, 11140–11144. [Google Scholar] [CrossRef]
- Dagkessamanskaia, A.; Moscoso, M.; Hénard, V.; Guiral, S.; Overweg, K.; Reuter, M.; Martin, B.; Wells, J.; Claverys, J.-P. Interconnection of competence, stress and CiaR regulons in Streptococcus pneumoniae: Competence triggers stationary phase autolysis of ciaR mutant cells. Mol. Microbiol. 2004, 51, 1071–1086. [Google Scholar] [CrossRef] [PubMed]
- Peterson, S.N.; Sung, C.K.; Cline, R.; Desai, B.V.; Snesrud, E.C.; Luo, P.; Walling, J.; Li, H.; Mintz, M.; Tsegaye, G.; et al. Identification of competence pheromone responsive genes in Streptococcus pneumoniae by use of DNA microarrays. Mol. Microbiol. 2004, 51, 1051–1070. [Google Scholar] [CrossRef] [PubMed]
- Slager, J.; Aprianto, R.; Veening, J.-W. Refining the Pneumococcal Competence Regulon by RNA Sequencing. J. Bacteriol. 2019, 201. [Google Scholar] [CrossRef]
- Liu, Y.; Zeng, Y.; Huang, Y.; Gu, L.; Wang, S.; Li, C.; Morrison, D.A.; Deng, H.; Zhang, J. HtrA-mediated selective degradation of DNA uptake apparatus accelerates termination of pneumococcal transformation. Mol. Microbiol. 2019, 112, 1308–1325. [Google Scholar] [CrossRef]
- Méjean, V.; Claverys, J.P. Use of a cloned DNA fragment to analyze the fate of donor DNA in transformation of Streptococcus pneumoniae. J. Bacteriol. 1984, 158, 1175–1178. [Google Scholar] [CrossRef] [PubMed]
- Bergé, M.J.; Kamgoué, A.; Martin, B.; Polard, P.; Campo, N.; Claverys, J.-P. Midcell Recruitment of the DNA Uptake and Virulence Nuclease, EndA, for Pneumococcal Transformation. PLoS Pathog. 2013, 9, e1003596. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Guild, W.R. Fractionated strands of bacterial deoxyribonucleic acid. 3. Transformation efficiencies and rates of phenotypic expression. J. Bacteriol. 1968, 96, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Louarn, J.-M.; Sicard, A.M. Identical transformability of both strands of recipient DNA in Diplococcus pneumoniae. Biochem. Biophys. Res. Commun. 1969, 36, 101–109. [Google Scholar] [CrossRef]
- Kurushima, J.; Campo, N.; van Raaphorst, R.; Cerckel, G.; Polard, P.; Veening, J.-W. Unbiased homeologous recombination during pneumococcal transformation allows for multiple chromosomal integration events. BioRxiv 2020. [Google Scholar] [CrossRef]
- Martin, B.; Granadel, C.; Campo, N.; Hénard, V.; Prudhomme, M.; Claverys, J.-P. Expression and maintenance of ComD-ComE, the two-component signal-transduction system that controls competence of Streptococcus pneumoniae. Mol. Microbiol. 2010, 75, 1513–1528. [Google Scholar] [CrossRef]
- Slager, J.; Kjos, M.; Attaiech, L.; Veening, J.-W. Antibiotic-Induced Replication Stress Triggers Bacterial Competence by Increasing Gene Dosage near the Origin. Cell 2014, 157, 395–406. [Google Scholar] [CrossRef]
- Moreno-Gámez, S.; Sorg, R.A.; Domenech, A.; Kjos, M.; Weissing, F.J.; van Doorn, G.S.; Veening, J.-W. Quorum sensing integrates environmental cues, cell density and cell history to control bacterial competence. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Domenech, A.; Slager, J.; Veening, J.-W. Antibiotic-Induced Cell Chaining Triggers Pneumococcal Competence by Reshaping Quorum Sensing to Autocrine-Like Signaling. Cell Rep. 2018, 25, 2390–2400. [Google Scholar] [CrossRef]
- Kjos, M.; Miller, E.; Slager, J.; Lake, F.B.; Gericke, O.; Roberts, I.S.; Rozen, D.E.; Veening, J.-W. Expression of Streptococcus pneumoniae Bacteriocins Is Induced by Antibiotics via Regulatory Interplay with the Competence System. PLOS Pathog. 2016, 12, e1005422. [Google Scholar] [CrossRef]
- Bergé, M.J.; Mercy, C.; Mortier-Barrière, I.; VanNieuwenhze, M.S.; Brun, Y.V.; Grangeasse, C.; Polard, P.; Campo, N. A programmed cell division delay preserves genome integrity during natural genetic transformation in Streptococcus pneumoniae. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, J.C.; Claverys, J.P.; Sicard, A.M. Donor deoxyribonucleic acid length and marker effect in pneumococcal transformation. J. Bacteriol. 1979, 138, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Mortier-Barrière, I.; Campo, N.; Bergé, M.A.; Prudhomme, M.; Polard, P. Natural Genetic Transformation: A Direct Route to Easy Insertion of Chimeric Genes into the Pneumococcal Chromosome. In Streptococcus Pneumoniae; Iovino, F., Ed.; Springer: New York, NY, USA, 2019; Volume 1968, pp. 63–78. ISBN 978-1-4939-9198-3. [Google Scholar]
- Martin, B.; Prudhomme, M.; Alloing, G.; Granadel, C.; Claverys, J.P. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol. Microbiol. 2000, 38, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Mirouze, N.; Bergé, M.A.; Soulet, A.-L.; Mortier-Barrière, I.; Quentin, Y.; Fichant, G.; Granadel, C.; Noirot-Gros, M.-F.; Noirot, P.; Polard, P.; et al. Direct involvement of DprA, the transformation-dedicated RecA loader, in the shut-off of pneumococcal competence. Proc. Natl. Acad. Sci. USA 2013, 110, E1035–E1044. [Google Scholar] [CrossRef] [PubMed]
- Mehta, G.; Mehta, K.; Sud, D.; Song, J.W.; Bersano-Begey, T.; Futai, N.; Heo, Y.S.; Mycek, M.-A.; Linderman, J.J.; Takayama, S. Quantitative measurement and control of oxygen levels in microfluidic poly(dimethylsiloxane) bioreactors during cell culture. Biomed. Microdevices 2007, 9, 123–134. [Google Scholar] [CrossRef]
- Taniai, H.; Iida, K.-i.; Seki, M.; Saito, M.; Shiota, S.; Nakayama, H.; Yoshida, S.-i. Concerted Action of Lactate Oxidase and Pyruvate Oxidase in Aerobic Growth of Streptococcus pneumoniae: Role of Lactate as an Energy Source. J. Bacteriol. 2008, 190, 3572–3579. [Google Scholar] [CrossRef]
- Johnsborg, O.; Kristiansen, P.E.; Blomqvist, T.; Havarstein, L.S. A Hydrophobic Patch in the Competence-Stimulating Peptide, a Pneumococcal Competence Pheromone, Is Essential for Specificity and Biological Activity. J. Bacteriol. 2006, 188, 1744–1749. [Google Scholar] [CrossRef][Green Version]
- Yang, Y.; Cornilescu, G.; Tal-Gan, Y. Structural Characterization of Competence-Stimulating Peptide Analogues Reveals Key Features for ComD1 and ComD2 Receptor Binding in Streptococcus pneumoniae. Biochemistry 2018, 57, 5359–5369. [Google Scholar] [CrossRef]
- Lee, P.; Gaige, T.; Hung, P. Microfluidic Systems for Live Cell Imaging. In Methods in Cell Biology; Elsevier: Amsterdam, The Netherlands, 2011; Volume 102, pp. 77–103. ISBN 978-0-12-374912-3. [Google Scholar]
- Balleza, E.; Kim, J.M.; Cluzel, P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat. Methods 2018, 15, 47–51. [Google Scholar] [CrossRef]
- Cranfill, P.J.; Sell, B.R.; Baird, M.A.; Allen, J.R.; Lavagnino, Z.; de Gruiter, H.M.; Kremers, G.-J.; Davidson, M.W.; Ustione, A.; Piston, D.W. Quantitative assessment of fluorescent proteins. Nat. Methods 2016, 13, 557–562. [Google Scholar] [CrossRef]
- Martin, B.; Soulet, A.-L.; Mirouze, N.; Prudhomme, M.; Mortier-Barrière, I.; Granadel, C.; Noirot-Gros, M.-F.; Noirot, P.; Polard, P.; Claverys, J.-P. ComE/ComE∼P interplay dictates activation or extinction status of pneumococcal X-state (competence): Dual control of early com promoters by ComE. Mol. Microbiol. 2013, 87, 394–411. [Google Scholar] [CrossRef] [PubMed]
- Ephrussi-Taylor, H. Appearance of Streptomycin Resistance Following the Uptake of Transforming Deoxyribonucleic Acid in Pneumococcus. Nature 1962, 196, 748–752. [Google Scholar] [CrossRef]
- Ephrussi-Taylor, H. Genetic recombination in DNA-induced transformation of Pneumococcus. IV. The pattern of transmission and phenotypic expression of high and low-efficiency donor sites in the amiA locus. Genetics 1966, 54, 211–222. [Google Scholar] [PubMed]
- Dalia, A.B.; Dalia, T.N. Spatiotemporal Analysis of DNA Integration during Natural Transformation Reveals a Mode of Nongenetic Inheritance in Bacteria. Cell 2019, 179, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Wehrens, M.; Ershov, D.; Rozendaal, R.; Walker, N.; Schultz, D.; Kishony, R.; Levin, P.A.; Tans, S.J. Size Laws and Division Ring Dynamics in Filamentous Escherichia coli cells. Curr. Biol. 2018, 28, 972–979. [Google Scholar] [CrossRef]
- Prudhomme, M.; Attaiech, L.; Sanchez, G.; Martin, B.; Claverys, J.-P. Antibiotic Stress Induces Genetic Transformability in the Human Pathogen Streptococcus pneumoniae. Science 2006, 313, 89–92. [Google Scholar] [CrossRef]
- Charpentier, X.; Polard, P.; Claverys, J.-P. Induction of competence for genetic transformation by antibiotics: Convergent evolution of stress responses in distant bacterial species lacking SOS? Curr. Opin. Microbiol. 2012, 15, 570–576. [Google Scholar] [CrossRef]
- Taheri-Araghi, S.; Brown, S.D.; Sauls, J.T.; McIntosh, D.B.; Jun, S. Single-Cell Physiology. Annu. Rev. Biophys. 2015, 44, 123–142. [Google Scholar] [CrossRef]
- Trojanowski, D.; Kołodziej, M.; Hołówka, J.; Müller, R.; Zakrzewska-Czerwińska, J. Watching DNA Replication Inhibitors in Action: Exploiting Time-Lapse Microfluidic Microscopy as a Tool for Target-Drug Interaction Studies in Mycobacterium. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mortier-Barrière, I.; Polard, P.; Campo, N. Direct Visualization of Horizontal Gene Transfer by Transformation in Live Pneumococcal Cells Using Microfluidics. Genes 2020, 11, 675. https://doi.org/10.3390/genes11060675
Mortier-Barrière I, Polard P, Campo N. Direct Visualization of Horizontal Gene Transfer by Transformation in Live Pneumococcal Cells Using Microfluidics. Genes. 2020; 11(6):675. https://doi.org/10.3390/genes11060675
Chicago/Turabian StyleMortier-Barrière, Isabelle, Patrice Polard, and Nathalie Campo. 2020. "Direct Visualization of Horizontal Gene Transfer by Transformation in Live Pneumococcal Cells Using Microfluidics" Genes 11, no. 6: 675. https://doi.org/10.3390/genes11060675
APA StyleMortier-Barrière, I., Polard, P., & Campo, N. (2020). Direct Visualization of Horizontal Gene Transfer by Transformation in Live Pneumococcal Cells Using Microfluidics. Genes, 11(6), 675. https://doi.org/10.3390/genes11060675