Conservation Assessment of the State Goat Farms by Using SNP Genotyping Data


Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Sampling, Genotyping and Quality Control
2.3. Genomic Relationship Analysis, and Population Structure
2.4. Runs of Homozygosity Calling Option and Estimation of Inbreeding
2.5. Effective Population Size (Ne)
3. Results
3.1. Sample and SNP Filtration
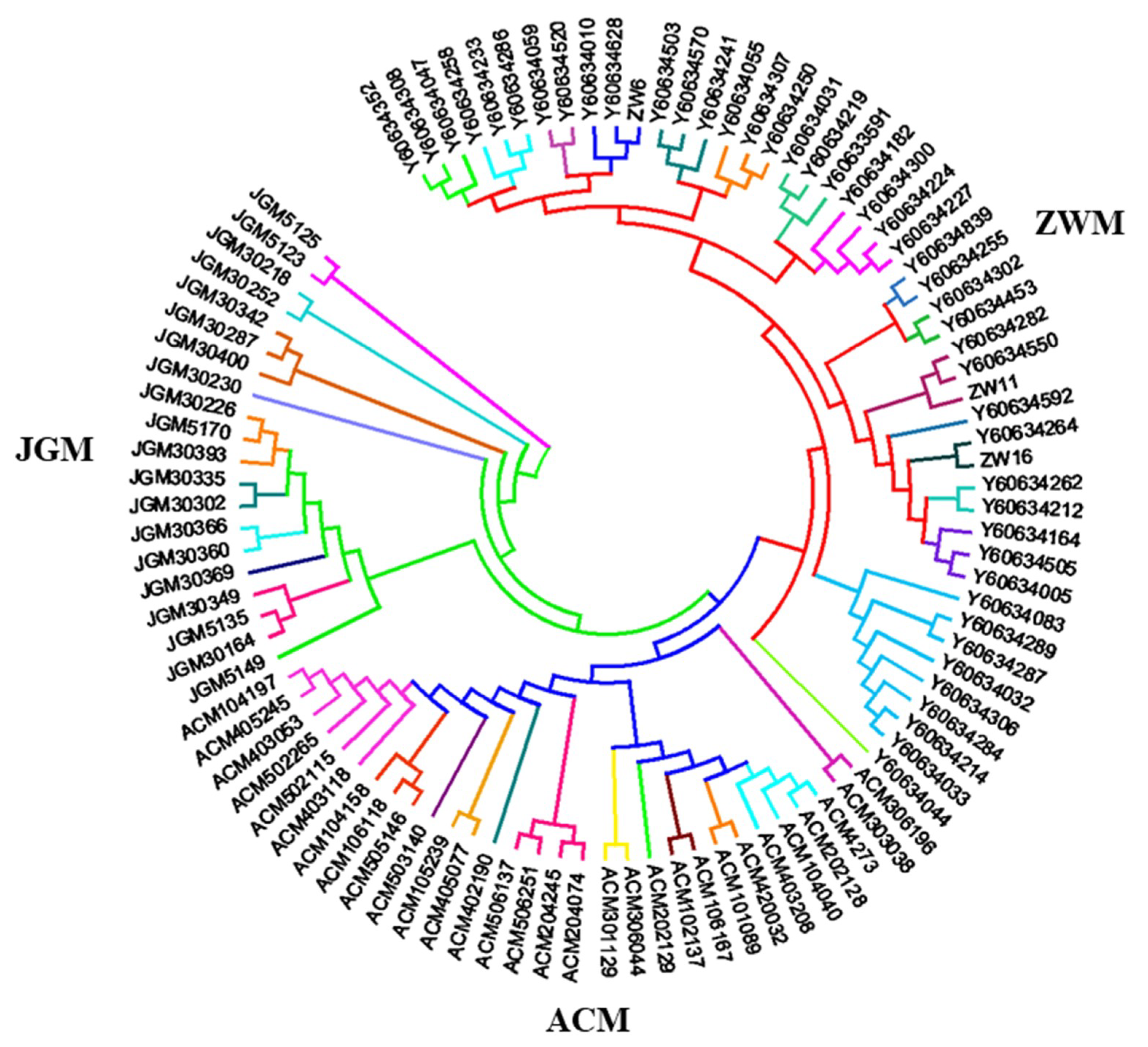
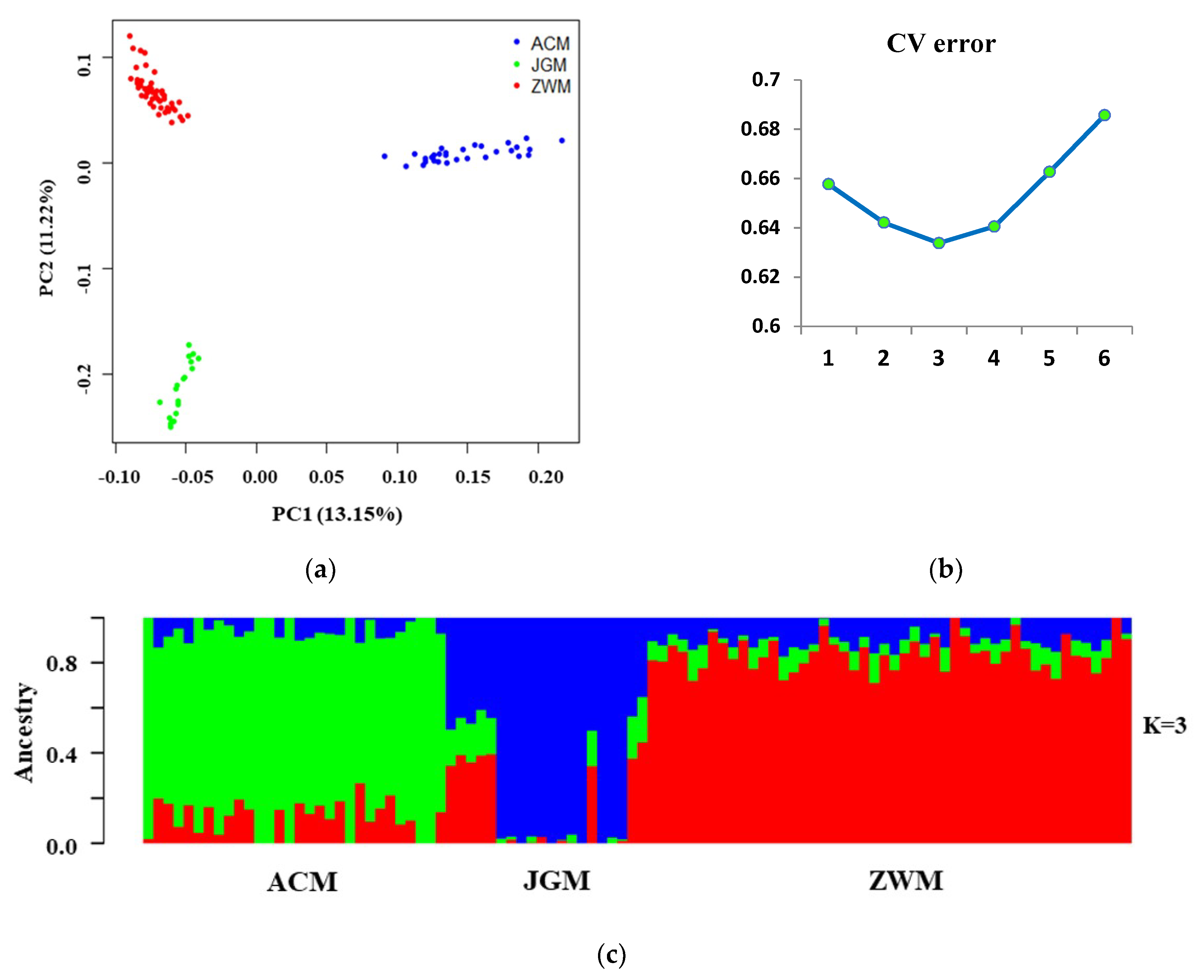
3.2. Genomic Relationship Analysis, and Population Structure
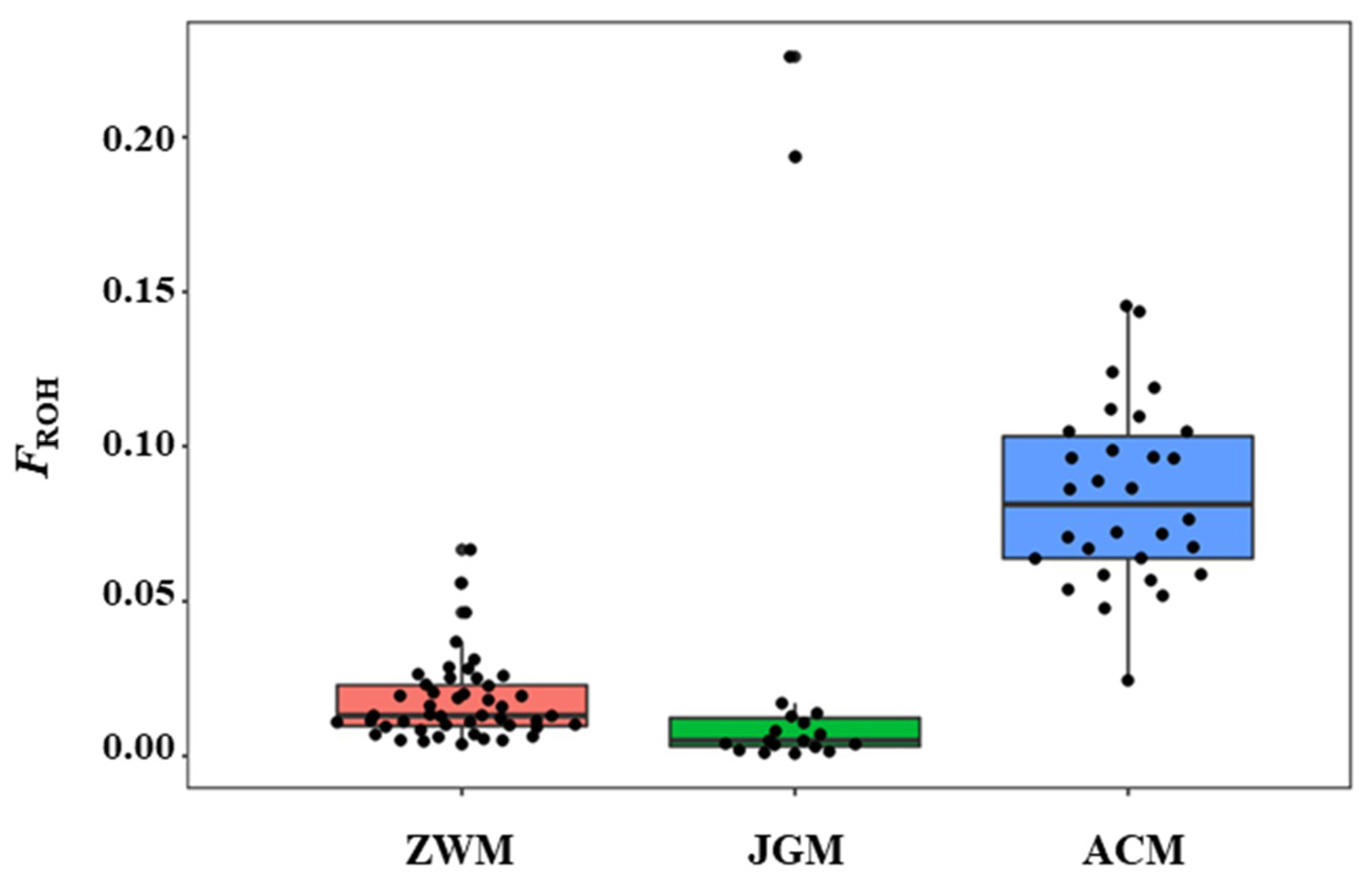
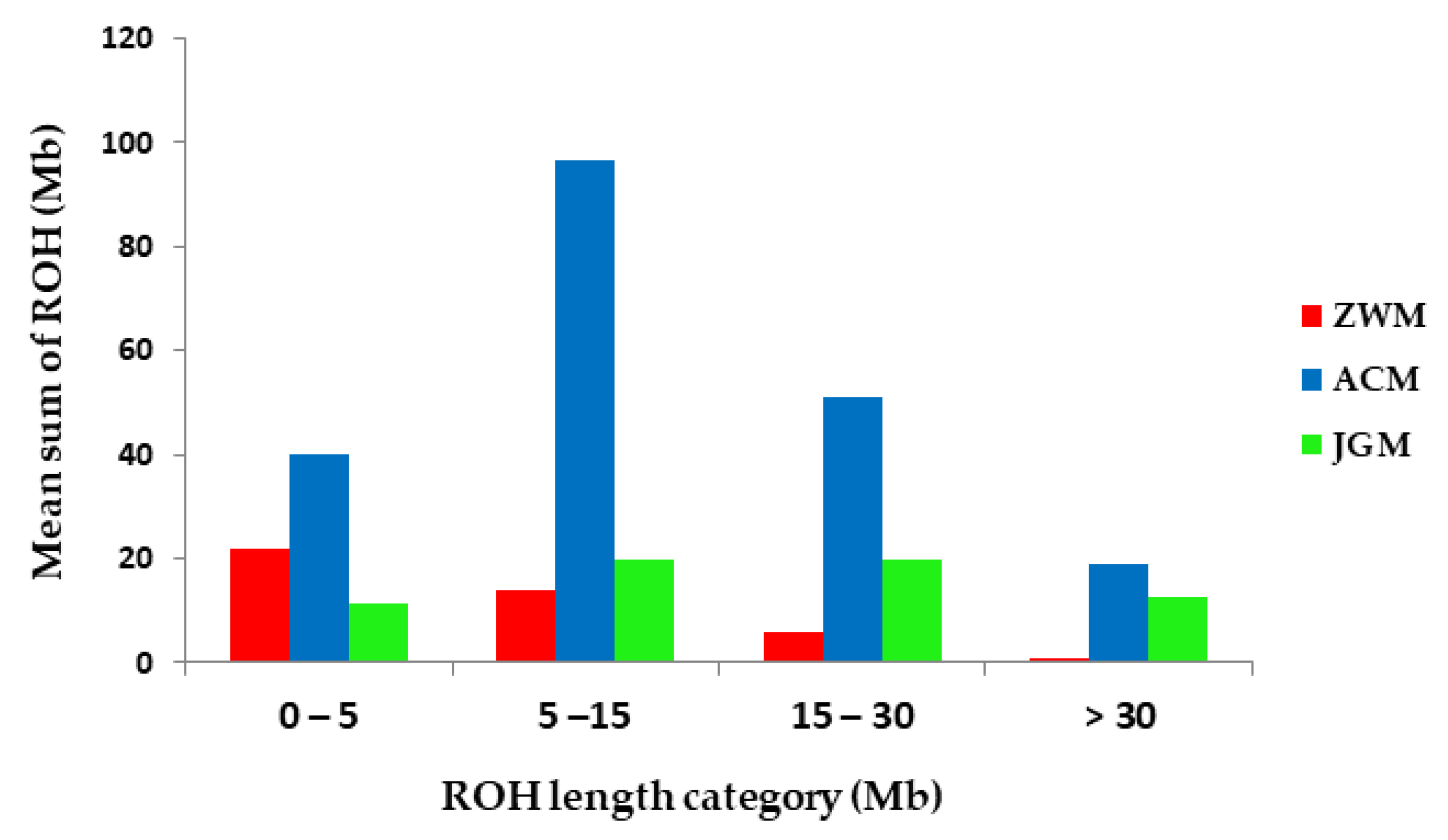
3.3. Genetic Diversity within Breeds
3.4. Effective Population Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fatima, S. Study of Genetic Variability Among Gohilwadi, Surti and Zalawadi Goats Using Microsatellite Analysis. Master’s Thesis, Anand Agricultural University, Anand, India, 2006. [Google Scholar]
- Gorbach, D.; Makgahlela, M.; Reecy, J.; Kemp, S.J.; Baltenweck, I.; Ouma, R.; Mwai, O.; Marshall, K.; Murdoch, B.; Moore, S.; et al. Use of SNP genotyping to determine pedigree and breed composition of dairy cattle in Kenya. J. Anim. Breed. Genet. 2010, 127, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.C.; Visscher, P.M.; Goddard, M.E. Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data. Genetics 2011, 189, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Calus, M.P.; Guldbrandtsen, B.; Lund, M.S.; Sahana, G. Estimation of inbreeding using pedigree, 50k SNP chip genotypes and full sequence data in three cattle breeds. BMC Genet. 2015, 16, 88. [Google Scholar] [CrossRef] [PubMed]
- Scraggs, E.; Zanella, R.; Wojtowicz, A.; Taylor, J.; Gaskins, C.; Reeves, J.; de Avila, J.; Neibergs, H. Estimation of inbreeding and effective population size of full-blood wagyu cattle registered with the American Wagyu Cattle Association. J. Anim. Breed. Genet. 2014, 131, 3–10. [Google Scholar] [CrossRef]
- Saura, M.; Fernández, A.; Varona, L.; Fernández, A.I.; de Cara, M.Á.R.; Barragán, C.; Villanueva, B.J.G.S.E. Detecting inbreeding depression for reproductive traits in Iberian pigs using genome-wide data. Genet. Sel. Evol. 2015, 47, 1. [Google Scholar] [CrossRef]
- Xie, R.; Shi, L.; Liu, J.; Deng, T.; Wang, L.; Liu, Y.; Zhao, F. Genome-Wide scan for runs of homozygosity identifies candidate genes in three pig breeds. Animals 2019, 9, 518. [Google Scholar] [CrossRef]
- Howrigan, D.P.; Simonson, M.A.; Keller, M.C. Detecting autozygosity through runs of homozygosity: A comparison of three autozygosity detection algorithms. BMC Genom. 2011, 12, 460. [Google Scholar] [CrossRef]
- Bosse, M.; Megens, H.-J.; Madsen, O.; Paudel, Y.; Frantz, L.A.; Schook, L.B.; Crooijmans, R.P.; Groenen, M.A. Regions of homozygosity in the porcine genome: Consequence of demography and the recombination landscape. PLoS Genet. 2012, 8, e1003100. [Google Scholar] [CrossRef]
- Onzima, R.B.; Upadhyay, M.R.; Doekes, H.P.; Brito, L.; Bosse, M.; Kanis, E.; Groenen, M.A.; Crooijmans, R.P. Genome-wide characterization of selection signatures and runs of homozygosity in Ugandan goat breeds. Front. Genet. 2018, 9, 318. [Google Scholar] [CrossRef]
- Kirin, M.; McQuillan, R.; Franklin, C.S.; Campbell, H.; McKeigue, P.M.; Wilson, J.F.J. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 2010, 5, e13996. [Google Scholar] [CrossRef]
- Shin, D.-H.; Cho, K.-H.; Park, K.-D.; Lee, H.-J.; Kim, H. Accurate estimation of effective population size in the Korean dairy cattle based on linkage disequilibrium corrected by genomic relationship matrix. Asian-Australas. J. Anim. Sci. 2013, 26, 1672. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Flury, C.; Tapio, M.; Sonstegard, T.; Drögemüller, C.; Leeb, T.; Simianer, H.; Hanotte, O.; Rieder, S. Effective population size of an indigenous Swiss cattle breed estimated from linkage disequilibrium. J. Anim. Breed. Genet. 2010, 127, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Peripolli, E.; Munari, D.; Silva, M.; Lima, A.; Irgang, R.; Baldi, F. Runs of homozygosity: Current knowledge and applications in livestock. Anim. Genet. 2017, 48, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; De Bakker, P.I.; Daly, M.J. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Islam, R.; Li, Y.; Liu, X.; Berihulay, H.; Abied, A.; Gebreselassie, G.; Ma, Q.; Ma, Y. Genome-Wide runs of homozygosity, effective population size, and detection of positive selection signatures in six Chinese Goat Breeds. Genes 2019, 10, 938. [Google Scholar] [CrossRef]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Silio, L.; Rodriguez, M.C.; Fernandez, A.; Barragan, C.; Benitez, R.; Ovilo, C.; Fernandez, A.I. Measuring inbreeding and inbreeding depression on pig growth from pedigree or SNP-derived metrics. Anim. Breed. Genet. 2013, 130, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Sved, J. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Hayes, B.J.; Visscher, P.M.; McPartlan, H.C.; Goddard, M.E. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 2003, 13, 635–643. [Google Scholar] [CrossRef]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of selection signature provides a glimpse into important economic traits in goats (Capra hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Li, X.; Han, W.; Yang, K.; Wang, H.; Zhang, Y.; Su, R.; Liu, Z.; Wang, R.; et al. High-throughput sequencing of hair follicle development-related micrornas in cashmere goat at various fetal periods. Saudi J. Biol. Sci. 2018, 25, 1494–1508. [Google Scholar] [CrossRef]
- Kumar, C.; Song, S.; Dewani, P.; Kumar, M.; Parkash, O.; Ma, Y.; Malhi, K.K.; Yang, N.; Mwacharo, J.M.; He, X.; et al. Population structure, genetic diversity and selection signatures within seven indigenous Pakistani goat populations. Anim. Genet. 2018, 49, 592–604. [Google Scholar] [CrossRef]
- Mdladla, K.; Dzomba, E.F.; Huson, H.J.; Muchadeyi, F.C. Population genomic structure and linkage disequilibrium analysis of South African goat breeds using genome-wide SNP data. Anim. Genet. 2016, 47, 471–482. [Google Scholar] [CrossRef]
- Tosser-Klopp, G.; Bardou, P.; Bouchez, O.; Cabau, C.; Crooijmans, R.; Dong, Y.; Donnadieu-Tonon, C.; Eggen, A.; Heuven, H.C.; Jamli, S.; et al. Design and characterization of a 52K SNP chip for goats. PLoS ONE 2014, 9, e86227. [Google Scholar] [CrossRef]
- Burren, A.; Neuditschko, M.; Signer-Hasler, H.; Frischknecht, M.; Reber, I.; Menzi, F.; Drogemuller, C.; Flury, C. Genetic diversity analyses reveal first insights into breed-specific selection signatures within Swiss goat breeds. Anim. Genet. 2016, 47, 727–739. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.; Ventura, R.V.; Baldi, F.; da Silva, M. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Tolone, M.; Sardina, M.T.; Sottile, G.; Sutera, A.M.; Di Gerlando, R.; Portolano, B. Genome-wide scan for runs of homozygosity identifies potential candidate genes associated with local adaptation in Valle del Belice sheep. Genet. Sel. Evol. 2017, 49, 84. [Google Scholar] [CrossRef] [PubMed]
- Visser, C.; Lashmar, S.F.; Van Marle-Koster, E.; Poli, M.A.; Allain, D. Genetic Diversity and population structure in South African, French and Argentinian Angora Goats from genome-wide SNP Data. PLoS ONE 2016, 11, e0154353. [Google Scholar] [CrossRef]
- Meuwissen, T. Genetic management of small populations: A review. Acta Agric. Scand. Sect. A Anim. Sci. 2009, 59, 71–79. [Google Scholar] [CrossRef]
- Brito, L.F.; Jafarikia, M.; Grossi, D.A.; Kijas, J.W.; Porto-Neto, L.R.; Ventura, R.V.; Salgorzaei, M.; Schenkel, F.S. Characterization of linkage disequilibrium, consistency of gametic phase and admixture in Australian and Canadian goats. BMC Genet. 2015, 16, 67. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Sample Size | Location | Utility, Demography and Breeding Practices | Agro-Ecology and Raising Methods | Pop. Trend |
|---|---|---|---|---|---|
| Zhongwei male (ZWM) | 48 | Zhongwei City, Ningxia Hui Autonomous Region | The Chinese Zhongwei goat is used primarily for the production of kid pelts. Total population was 30,000 in 2006. It is an excellent local variety after natural and artificial selection. | Av. Altitude 1200 m. Semi-pasture land, semi-dry land, mainly stall-feeding reared. | Negative |
| Jining Grey male (JGM) | 20 | Jining City, Shandong Province, China | They are fast maturing and very prolific, with an average kidding rate of 283%. Total population was 430,000 in 2006. Bred in the local natural environment, after many years of selection their adaptability is high, high fecundity rate. | Av. Altitude 45 m. Agricultural land, wetland, grazing in mountain area in summer and stall feeding in winter | Stable |
| Arbas Cashmere male (ACM) | 30 | Erdos City, Inner Mongolia Autonomous Region, China | It is well known for its excellent quality cashmere fiber. Total population was 4.3 million in 2006. After long-term natural and artificial selection under difficult ecological conditions, the cashmere and meat varieties are adapted to the local conditions. | Av. Altitude 1500 m. Semi-pasture land, semi-dry land. Mainly grazing practices. | Positive |
| Parameters | Excluded SNPs | SNP Remained |
|---|---|---|
| Total number of SNPs | 53,347 | |
| SNPs removed due to unrecognized position | 2773 | 50,574 |
| SNP Call frequency (call rate) (<0.95) | 1956 | 48,618 |
| Minor allele frequency (<0.05) | 3425 | 45,193 |
| Hardy-Weinberg equilibrium (p < 1 × 10−5) | 152 | 45,041 |
| Total SNPs | 8306 | 45,041 |
| Breed | n | % PN | HO | HE | FROH | FHOM | R (FROH, FHOM) |
|---|---|---|---|---|---|---|---|
| ZWM | 48 | 95.2 | 0.385 ± 0.20 | 0.386 ± 0.19 | 0.017 | 0.021 | 0.92 |
| ACM | 30 | 91.8 | 0.367 ± 0.19 | 0.384 ± 0.20 | 0.086 | 0.074 | 0.99 |
| JGM | 20 | 96.2 | 0.401 ± 0.20 | 0.391 ± 0.20 | 0.028 | −0.010 | 0.96 |
| Overall | 98 | 96.8 | 0.385 ± 0.19 | 0.397 ± 0.20 | 0.040 | 0.032 | 0.92 |
| Breed | 0–5 Mb | 5–15 Mb | 15–30 Mb | >30 Mb | Total |
|---|---|---|---|---|---|
| ZWM | 0.92 | 0.59 | 0.25 | 0.03 | 1.79 |
| ACM | 1.67 | 4.03 | 2.13 | 0.79 | 8.62 |
| JGM | 0.47 | 0.83 | 0.83 | 0.53 | 2.66 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, R.; Liu, Z.; Li, Y.; Jiang, L.; Ma, Y. Conservation Assessment of the State Goat Farms by Using SNP Genotyping Data. Genes 2020, 11, 652. https://doi.org/10.3390/genes11060652
Islam R, Liu Z, Li Y, Jiang L, Ma Y. Conservation Assessment of the State Goat Farms by Using SNP Genotyping Data. Genes. 2020; 11(6):652. https://doi.org/10.3390/genes11060652
Chicago/Turabian StyleIslam, Rabiul, Zhangfa Liu, Yefang Li, Lin Jiang, and Yuehui Ma. 2020. "Conservation Assessment of the State Goat Farms by Using SNP Genotyping Data" Genes 11, no. 6: 652. https://doi.org/10.3390/genes11060652
APA StyleIslam, R., Liu, Z., Li, Y., Jiang, L., & Ma, Y. (2020). Conservation Assessment of the State Goat Farms by Using SNP Genotyping Data. Genes, 11(6), 652. https://doi.org/10.3390/genes11060652