Association of Common Genetic Variants in the CPSF7 and SDHAF2 Genes with Canine Idiopathic Pulmonary Fibrosis in the West Highland White Terrier



Abstract
1. Introduction
2. Methods
2.1. Sample Collection
2.2. DNA Extraction and Whole Genome Sequencing
2.3. Data Analysis
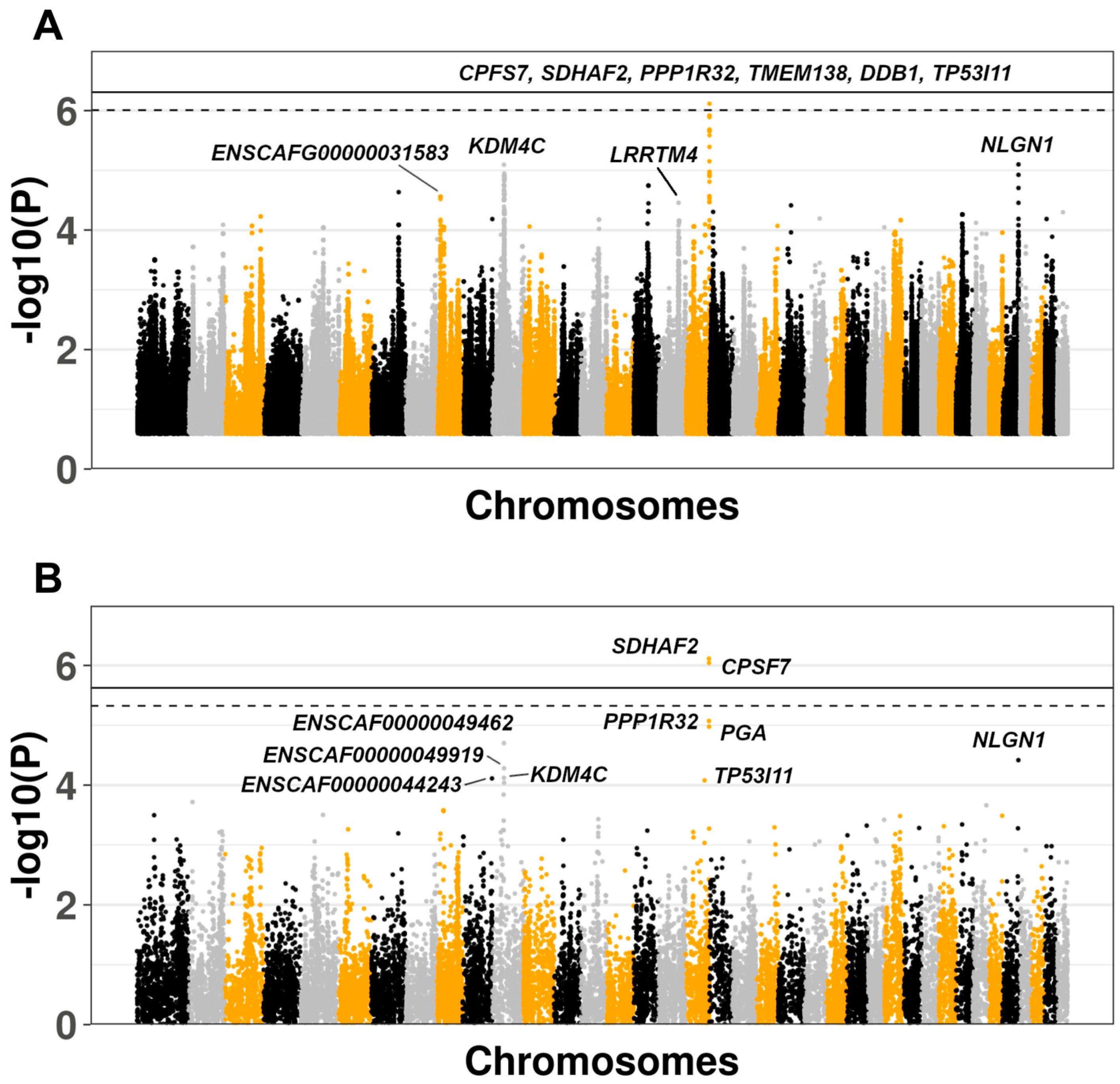
3. Results
3.1. Filtering and Quality Controls
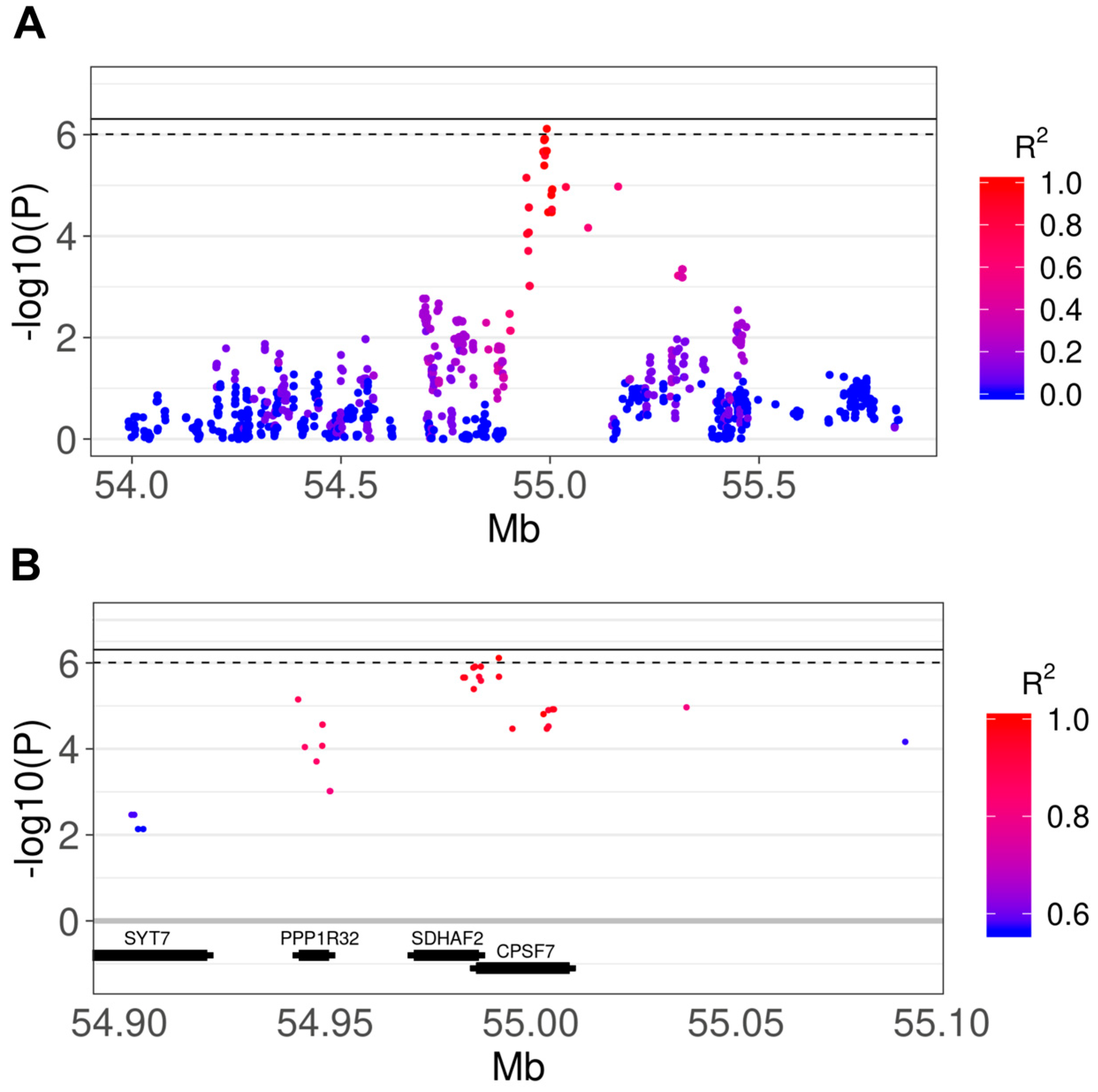
3.2. Genome-Wide Association Study
3.3. Comparison of Results With Human IPF GWAS and Canine SNP Reference Data
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Heikkilä, H.P.; Lappalainen, A.K.; Day, M.J.; Clercx, C.; Rajamäki, M.M. Clinical, bronchoscopic, histopathologic, diagnostic imaging, and arterial oxygenation findings in west highland white terriers with idiopathic pulmonary fibrosis. J. Vet. Intern. Med. 2011, 25, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Clercx, C.; Fastrès, A.; Roels, E. Idiopathic pulmonary fibrosis in West Highland white terriers: An update. Vet. J. 2018, 242, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä-Laurila, H.P.; Rajamäki, M.M. Idiopathic pulmonary fibrosis in west highland white terriers. Vet. Clin. N. Am. Small Anim. Pract. 2014, 44, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Daccord, C.; Maher, T.M. Recent advances in understanding idiopathic pulmonary fibrosis. F1000Research 2016, 5, 1046. [Google Scholar] [CrossRef] [PubMed]
- Coward, W.R.; Saini, G.; Jenkins, G. The pathogenesis of idiopathic pulmonary fibrosis. Ther. Adv. Respir. Dis. 2010, 4, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Lilja-Maula, L.; Syrjä, P.; Laurila, H.P.; Sutinen, E.; Palviainen, M.; Ritvos, O.; Koli, K.; Rajamäki, M.M.; Myllärniemi, M. Upregulation of alveolar levels of activin B, but not activin A, in lungs of west highland white terriers with idiopathic pulmonary fibrosis and diffuse alveolar damage. J. Comp. Pathol. 2015, 152, 192–200. [Google Scholar] [CrossRef]
- Krafft, E.; Lybaert, P.; Roels, E.; Laurila, H.P.; Rajamäki, M.M.; Farnir, F.; Myllärniemi, M.; Day, M.J.; Mc Entee, K.; Clercx, C. Transforming Growth Factor Beta 1 Activation, Storage, and Signaling Pathways in Idiopathic Pulmonary Fibrosis in Dogs. J. Vet. Intern. Med. 2014, 28, 1666–1675. [Google Scholar] [CrossRef]
- Krafft, E.; Laurila, H.P.; Peters, I.R.; Bureau, F.; Peeters, D.; Day, M.J.; Rajamäki, M.M.; Clercx, C. Analysis of gene expression in canine idiopathic pulmonary fibrosis. Vet. J. 2013, 198, 479–486. [Google Scholar] [CrossRef]
- Krafft, E.; Heikkilä, H.P.; Jespers, P.; Peeters, D.; Day, M.J.; Rajamäki, M.M.; Mc Entee, K.; Clercx, C. Serum and Bronchoalveolar Lavage Fluid Endothelin-1 Concentrations as Diagnostic Biomarkers of Canine Idiopathic Pulmonary Fibrosis. J. Vet. Intern. Med. 2011, 25, 990–996. [Google Scholar] [CrossRef]
- Noth, I.; Zhang, Y.; Ma, S.F.; Flores, C.; Barber, M.; Huang, Y.; Broderick, S.M.; Wade, M.S.; Hysi, P.; Scuirba, J.; et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: A genome-wide association study. Lancet Respir. Med. 2013, 1, 309–317. [Google Scholar] [CrossRef]
- Fingerlin, T.E.; Murphy, E.; Zhang, W.; Peljto, A.L.; Brown, K.K.; Steele, M.P.; Loyd, J.E.; Cosgrove, G.P.; Lynch, D.; Groshong, S.; et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat. Genet. 2013, 45, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Mushiroda, T.; Wattanapokayakit, S.; Takahashi, A.; Nukiwa, T.; Kudoh, S.; Ogura, T.; Taniguchi, H.; Kubo, M.; Kamatani, N.; Nakamura, Y. A genome-wide association study identifies an association of a common variant in TERT with susceptibility to idiopathic pulmonary fibrosis. J. Med. Genet. 2008, 45, 654–656. [Google Scholar] [CrossRef] [PubMed]
- Shearin, A.L.; Ostrander, E.A. Leading the way: Canine models of genomics and disease. Dis. Model. Mech. 2010, 3, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wayne, R.K.; Ostrander, E.A. Lessons learned from the dog genome. Trends Genet. 2007, 23, 557–567. [Google Scholar] [CrossRef]
- Lindblad-Toh, K.; Wade, C.M.; Mikkelsen, T.S.; Karlsson, E.K.; Jaffe, D.B.; Kamal, M.; Clamp, M.; Chang, J.L.; Kulbokas, E.J.; Zody, M.C.; et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 2005, 438, 803–819. [Google Scholar] [CrossRef]
- Ostrander, E.A.; Kruglyak, L. Unleashing the canine genome. Genome Res. 2000, 10, 1271–1274. [Google Scholar] [CrossRef][Green Version]
- Gallana, M.; Utsunomiya, Y.T.; Dolf, G.; Pintor Torrecilha, R.B.; Falbo, A.K.; Jagannathan, V.; Leeb, T.; Reichler, I.; Sölkner, J.; Schelling, C. Genome-wide association study and heritability estimate for ectopic ureters in Entlebucher mountain dogs. Anim. Genet. 2018, 49, 645–650. [Google Scholar] [CrossRef]
- Peiravan, A.; Bertolini, F.; Rothschild, M.F.; Simpson, K.W.; Jergens, A.E.; Allenspach, K.; Werling, D. Genome-wide association studies of inflammatory bowel disease in German shepherd dogs. PLoS ONE 2018, 13, e0200685. [Google Scholar] [CrossRef]
- Gast, A.C.; Metzger, J.; Tipold, A.; Distl, O. Genome-wide association study for hereditary ataxia in the Parson Russell Terrier and DNA-testing for ataxia-associated mutations in the Parson and Jack Russell Terrier. BMC Vet. Res. 2016, 12, 225. [Google Scholar] [CrossRef]
- Bianchi, M.; Dahlgren, S.; Massey, J.; Dietschi, E.; Kierczak, M.; Lund-Ziener, M.; Sundberg, K.; Thoresen, S.I.; Kämpe, O.; Andersson, G.; et al. A multi-breed genome-wide association analysis for canine Hypothyroidism identifies a shared major risk locus on CFA12. PLoS ONE 2015, 10, e0134720. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Stephens, M. Modelling linkage disequilibrium and identifying recombination hotspots using SNP data genetics. Genetics 2003, 165, 2213–2233. [Google Scholar] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.E.; Daly, M.J.; et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed-model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Stephens, M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat. Methods 2014, 11, 407–409. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef]
- Li, M.-X.; Gui, H.-S.; Kwan, J.S.H.; Sham, P.C. GATES: A rapid and powerful gene-based association test using extended Simes procedure. Am. J. Hum. Genet. 2011, 88, 283–293. [Google Scholar] [CrossRef]
- Allen, R.J.; Porte, J.; Braybrooke, R.; Flores, C.; Fingerlin, T.E.; Oldham, J.M.; Guillen-Guio, B.; Ma, S.F.; Okamoto, T.; John, A.E.; et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: A genome-wide association study. Lancet Respir. Med. 2017, 5, 869–880. [Google Scholar] [CrossRef]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Hayward, J.J.; White, M.E.; Boyle, M.; Shannon, L.M.; Casal, M.L.; Castelhano, M.G.; Center, S.A.; Meyers-Wallen, V.N.; Simpson, K.W.; Sutter, N.B.; et al. Imputation of canine genotype array data using 365 whole-genome sequences improves power of genome-wide association studies. PLOS Genet. 2019, 15, e1008003. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, L.; Zhou, Z.; Liu, Y.; Sun, J.; Zhang, X.; Pan, H.; Liu, S. SP1 induced long non-coding RNA LINC00958 overexpression facilitate cell proliferation, migration and invasion in lung adenocarcinoma via mediating miR-625-5p/CPSF7 axis. Cancer Cell Int. 2020, 20, 24. [Google Scholar] [CrossRef] [PubMed]
- Ballester, B.; Milara, J.; Cortijo, J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int. J. Mol. Sci. 2019, 20, 593. [Google Scholar] [CrossRef]
- Fang, S.; Zhang, D.; Weng, W.; Lv, X.; Zheng, L.; Chen, M.; Fan, X.; Mao, J.; Mao, C.; Ye, Y.; et al. CPSF7 regulates liver cancer growth and metastasis by facilitating WWP2-FL and targeting the WWP2/PTEN/AKT signaling pathway. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118624. [Google Scholar] [CrossRef] [PubMed]
- Bausch, B.; Schiavi, F.; Ni, Y.; Welander, J.; Patocs, A.; Ngeow, J.; Wellner, U.; Malinoc, A.; Taschin, E.; Barbon, G.; et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017, 3, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.D.; Harvey, R.N.; Darr, O.A.; Prince, M.E.; Bradford, C.R.; Wolf, G.T.; Else, T.; Basura, G.J. Head and neck paragangliomas: A two-decade institutional experience and algorithm for management. Laryngoscope Investig. Otolaryngol. 2017, 2, 380–389. [Google Scholar] [CrossRef]
- Corcoran, B.M.; Cobb, M.; Martin, M.W.S.; Dukes-McEwan, J.; French, A.; Luis Fuentes, V.; Boswood, A.; Rhind, S. Chronic pulmonary disease in West Highland white terriers. Vet. Rec. 1999, 144, 611–616. [Google Scholar] [CrossRef]
- Roels, E.; Fastrès, A.; Gommeren, K.; Saegerman, C.; Clercx, C. A questionnaire-based survey of owner-reported environment and care of West Highland white Terrier with or without idiopathic pulmonary fibrosis. In Proceedings of the 24th ECVIM-CA Congress, Mainz, Germany, 4–6 September 2014. [Google Scholar]



{kind=link}
{kind=link}
| Refsnp ID | CHR | BP | A1 | A2 | F_A | F_U | Depth (SD) | Beta | p | Adj p | Ensembl Gene ID | Gene Name | Consequence Type |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs22669389 | 18 | 54992254 | T | A | 0.704 | 0.333 | 0.451 ± 0.713 | 0.406 | 7.7 × 10−7 | 0.078 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs22647286 | 18 | 54987884 | C | T | 0.704 | 0.333 | 3.183 ± 2.875 | 0.394 | 1.2 × 10−6 | 0.124 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs851654341 | 18 | 54986491 | A | G | 0.704 | 0.333 | 2.324 ± 2.123 | 0.394 | 1.2 × 10−6 | 0.124 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs852097932 | 18 | 54986070 | A | G | 0.704 | 0.337 | 2.861 ± 2.209 | 0.394 | 1.3 × 10−6 | 0.131 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs22686152 | 18 | 54992285 | A | G | 0.704 | 0.345 | 0.732 ± 0.940 | 0.386 | 2.1 × 10−6 | 0.213 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs22647289 | 18 | 54987464 | G | T | 0.704 | 0.345 | 5.423 ± 3.702 | 0.391 | 2.1 × 10−6 | 0.214 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | 5’ UTR, u; 5’ UTR, u |
| rs850942449 | 18 | 54983627 | A | G | 0.704 | 0.345 | 2.831 ± 2.449 | 0.393 | 2.2 × 10−6 | 0.223 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| - | 18 | 54984004 | G | A | 0.704 | 0.345 | 0.887 ± 0.919 | 0.393 | 2.2 × 10−6 | 0.223 | ENSCAFG00000030303 | SDHAF2 | i |
| rs22647283 | 18 | 54987912 | C | T | 0.692 | 0.326 | 2.535 ± 2.709 | 0.390 | 2.6 × 10−6 | 0.263 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
| rs850871193 | 18 | 54986170 | C | T | 0.692 | 0.337 | 3.028 ± 2.646 | 0.387 | 4.1 × 10−6 | 0.413 | ENSCAFG00000030303; ENSCAFG00000016152 | SDHAF2; CPSF7 | u, i; u, i |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piras, I.S.; Bleul, C.; Siniard, A.; Wolfe, A.J.; De Both, M.D.; Hernandez, A.G.; Huentelman, M.J. Association of Common Genetic Variants in the CPSF7 and SDHAF2 Genes with Canine Idiopathic Pulmonary Fibrosis in the West Highland White Terrier. Genes 2020, 11, 609. https://doi.org/10.3390/genes11060609
Piras IS, Bleul C, Siniard A, Wolfe AJ, De Both MD, Hernandez AG, Huentelman MJ. Association of Common Genetic Variants in the CPSF7 and SDHAF2 Genes with Canine Idiopathic Pulmonary Fibrosis in the West Highland White Terrier. Genes. 2020; 11(6):609. https://doi.org/10.3390/genes11060609
Chicago/Turabian StylePiras, Ignazio S., Christiane Bleul, Ashley Siniard, Amanda J. Wolfe, Matthew D. De Both, Alvaro G. Hernandez, and Matthew J. Huentelman. 2020. "Association of Common Genetic Variants in the CPSF7 and SDHAF2 Genes with Canine Idiopathic Pulmonary Fibrosis in the West Highland White Terrier" Genes 11, no. 6: 609. https://doi.org/10.3390/genes11060609
APA StylePiras, I. S., Bleul, C., Siniard, A., Wolfe, A. J., De Both, M. D., Hernandez, A. G., & Huentelman, M. J. (2020). Association of Common Genetic Variants in the CPSF7 and SDHAF2 Genes with Canine Idiopathic Pulmonary Fibrosis in the West Highland White Terrier. Genes, 11(6), 609. https://doi.org/10.3390/genes11060609