Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain

 ,
,  ,
,  , , , , ,
, , , , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Genetic Analysis
2.3. Data Analysis and Interpretation
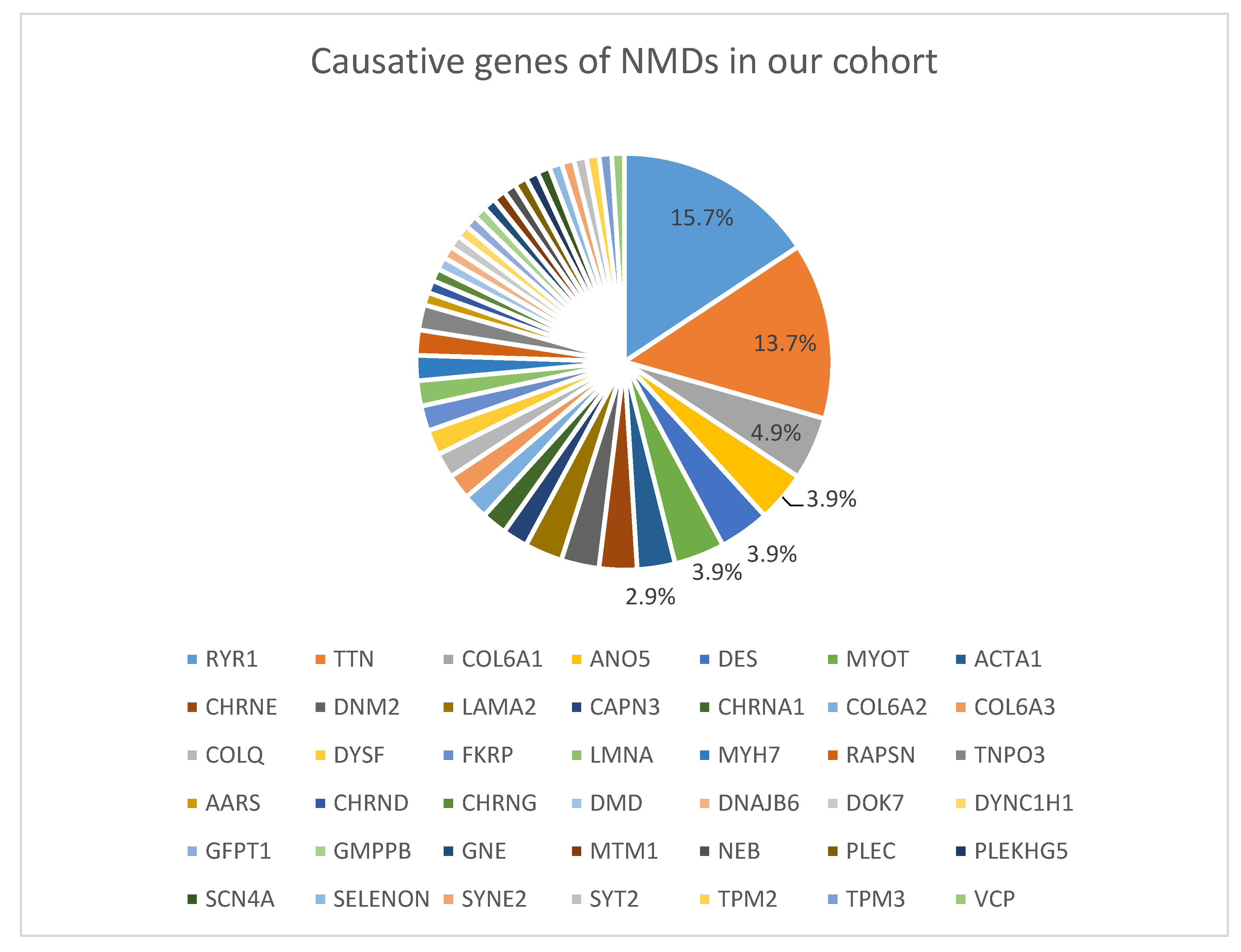
3. Results
3.1. Coverage and Sequencing Depths
3.2. Identified Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Available online: www.musclegenetable.fr (accessed on 10 May 2020).
- Kirschner, J.; Bonnemann, C.; Schorling, D. Congenital Muscular Dystrophies and Myopathies: An Overview and Update. Neuropediatrics 2017, 48, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Scoto, M.; Finkel, R.; Mercuri, E.; Muntoni, F. Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc. Health 2018, 2, 600–609. [Google Scholar] [CrossRef]
- Emery, A.E.H. Seminar: The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Sewry, C.A. Pathological defects in congenital myopathies. J. Muscle Res. Cell Motil. 2008, 29, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.-M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 420–434. [Google Scholar] [CrossRef]
- Finsterer, J. Congenital myasthenic syndromes. Lancet Neurol. 2019, 5, 1–22. [Google Scholar] [CrossRef]
- Nardin, R.; Johns, D.R. Mitochondrial dysfunction and neuromuscular disease. Muscle Nerve 2001, 24, 170–179. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Koutzaki, S.; Melvin, J.J. Mitochondrial Dysfunction in Neuromuscular Disorders. Semin. Pediatr. Neurol. 2013, 20, 202–215. [Google Scholar] [CrossRef]
- Nigro, V.; Savarese, M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr. Opin. Neurol. 2016, 29, 621–627. [Google Scholar] [CrossRef]
- Chae, J.H.; Vasta, V.; Cho, A.; Lim, B.C.; Zhang, Q.; Eun, S.-H.; Hahn, S.H. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J. Med Genet. 2015, 52, 208–216. [Google Scholar] [CrossRef]
- Tian, X.; Liang, W.-C.; Feng, Y.; Wang, J.; Zhang, V.W.; Chou, C.H.; Huang, H.-D.; Lam, C.W.; Hsu, Y.-Y.; Lin, T.-S.; et al. Expanding genotype/phenotype of neuromuscular diseases by comprehensive target capture/NGS. Neurol. Genet. 2015, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Ghaoui, R.; Cooper, S.T.; Lek, M.; Jones, K.; Corbett, A.; Reddel, S.W.; Needham, M.; Liang, C.; Waddell, L.B.; Nicholson, G.; et al. Use of Whole-Exome Sequencing for Diagnosis of Limb-Girdle Muscular Dystrophy. JAMA Neurol. 2015, 72, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Glaser, D.; Joshi, P.R.; Zierz, S.; Wenninger, S.; Schoser, B.; Deschauer, M. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. J. Neurol. 2016, 263, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Seong, M.-W.; Cho, A.; Park, H.; Seo, S.; Lim, B.; Seol, D.; Cho, S.; Park, S.S.; Chae, J. Clinical applications of next-generation sequencing-based gene panel in patients with muscular dystrophy: Korean experience. Clin. Genet. 2015, 89, 484–488. [Google Scholar] [CrossRef]
- Ankala, A.; Da Silva, C.; Gualandi, F.; Ferlini, A.; Bean, L.J.H.; Collins, C.; Tanner, A.K.; Hegde, M.R. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann. Neurol. 2014, 77, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kondo, E.; Urano, M.; Aoki, R.; Saito, K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J. Hum. Genet. 2016, 61, 931–942. [Google Scholar] [CrossRef]
- Richards, S.; on behalf of the ACMG Laboratory Quality Assurance Committee; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Hackman, P.; Marchand, S.; Sarparanta, J.; Vihola, A.; Penisson-Besnier, I.; Eymard, B.; Pardal-Fernández, J.M.; Hammouda, E.-H.; Richard, I.; Illa, I.; et al. Truncating mutations in C-terminal titin may cause more severe tibial muscular dystrophy (TMD). Neuromuscul. Disord. 2008, 18, 922–928. [Google Scholar] [CrossRef]
- Donger, C.; Krejci, E.; Serradell, A.P.; Eymard, B.; Bon, S.; Nicole, S.; Chateau, D.; Gary, F.; Fardeau, M.; Massoulié, J.; et al. Mutation in the Human Acetylcholinesterase-Associated Collagen Gene, COLQ, Is Responsible for Congenital Myasthenic Syndrome with End-Plate Acetylcholinesterase Deficiency (Type Ic). Am. J. Hum. Genet. 1998, 63, 967–975. [Google Scholar] [CrossRef]
- Selcen, D.; Milone, M.; Shen, X.-M.; Harper, C.M.; Stans, A.A.; Wieben, E.D.; Engel, A.G. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Ann. Neurol. 2008, 64, 71–87. [Google Scholar] [CrossRef]
- Todd, E.J.; Yau, K.S.; Ong, R.; Slee, J.; McGillivray, G.; Barnett, C.P.; Haliloglu, G.; Talim, B.; Akçören, Z.; Kariminejad, A.; et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet J. Rare Dis. 2015, 10, 148. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, A.; Mitsuhashi, S.; Miyata, N.; Nishino, I. Targeted massively parallel sequencing and histological assessment of skeletal muscles for the molecular diagnosis of inherited muscle disorders. J. Med Genet. 2016, 54, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Di Fruscio, G.; Torella, A.; Fiorillo, C.; Magri, F.; Fanin, M.; Ruggiero, L.; Ricci, G.; Astrea, G.; Passamano, L.; et al. The genetic basis of undiagnosed muscular dystrophies and myopathies: Results from 504 patients. Neurology 2016, 87, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Vasli, N.; Böhm, J.; Le Gras, S.; Muller, J.; Pizot, C.; Jost, B.; Echaniz-Laguna, A.; Laugel, V.; Tranchant, C.; Bernard, R.; et al. Next generation sequencing for molecular diagnosis of neuromuscular diseases. Acta Neuropathol. 2012, 124, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Savarese, M.; Di Fruscio, G.; Mutarelli, M.; Torella, A.; Magri, F.; Santorelli, F.M.; Comi, G.P.; Bruno, C.; Nigro, V. MotorPlex provides accurate variant detection across large muscle genes both in single myopathic patients and in pools of DNA samples. Acta Neuropathol. Commun. 2014, 2, 100. [Google Scholar] [CrossRef]
- Davis, M.; Haan, E.; Jungbluth, H.; Sewry, C.; North, K.; Muntoni, F.; Kuntzer, T.; Lamont, P.; Bankier, A.; Tomlinson, P.; et al. Principal mutation hotspot for central core disease and related myopathies in the C-terminal transmembrane region of the RYR1 gene. Neuromuscul. Disord. 2003, 13, 151–157. [Google Scholar] [CrossRef]
- Amburgey, K.; Bailey, A.; Hwang, J.H.; Tarnopolsky, M.A.; Bönnemann, C.G.; Medne, L.; Mathews, K.; Collins, J.; Daube, J.R.; Wellman, G.P.; et al. Genotype-phenotype correlations in recessive RYR1-related myopathies. Orphanet J. Rare Dis. 2013, 8, 117. [Google Scholar] [CrossRef]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef]
- Evilä, A.; Vihola, A.; Sarparanta, J.; Raheem, O.; Palmio, J.; Sandell, S.; Eymard, B.; Illa, I.; Rojas-Garcia, R.; Hankiewicz, K.; et al. Atypical phenotypes in titinopathies explained by second titin mutations. Ann. Neurol. 2014, 75, 230–240. [Google Scholar] [CrossRef]
- Cooper, S.T.; Kizana, Y.; Yates, J.D.; Lo, H.; Yang, N.; Wu, Z.H.; Alexander, I.E.; North, K. Dystrophinopathy carrier determination and detection of protein deficiencies in muscular dystrophy using lentiviral MyoD-forced myogenesis. Neuromuscul. Disord. 2007, 17, 276–284. [Google Scholar] [CrossRef]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Spina, S.; Van Laar, A.D.; Murrell, J.R.; Hamilton, R.L.; Kofler, J.K.; Epperson, F.; Farlow, M.R.; Lopez, O.L.; Quinlan, J.; DeKosky, S.T.; et al. Phenotypic variability in three families with valosin-containing protein mutation. Eur. J. Neurol. 2012, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Vissing, J.; Barresi, R.; Witting, N.; Van Ghelue, M.; Gammelgaard, L.; Bindoff, L.A.; Straub, V.; Lochmüller, H.; Hudson, J.; Wahl, C.M.; et al. A heterozygous 21-bp deletion inCAPN3causes dominantly inherited limb girdle muscular dystrophy. Brain 2016, 139, 2154–2163. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef]


{kind=link}
| AARS | CRYAB | KLHL40 | PREPL |
| ACTA1 | DAG1 | KLHL41 | PTPLA |
| ACVR1 | DES | KLHL9 | PTRF |
| AGRN | DMD | LAMA2 | RAPSN |
| ALG13 | DNAJB6 | LAMB2 | RYR1 |
| ALG14 | DNM2 | LAMP2 | SCN4A |
| ALG2 | DOK7 | LARGE | SEPN1 |
| ANO5 | DPAGT1 | LDB3 | SGCA |
| B3GALNT2 | DPM1 | LMNA | SGCB |
| B3GNT1 | DPM2 | LMOD3 | SGCD |
| BAG3 | DPM3 | LRP4 | SGCG |
| BIN1 | DYNC1H1 | MEGF10 | SGK196 |
| CAPN3 | DYSF | MSTN | SPEG |
| CAV3 | EMD | MTM1 | STIM1 |
| CCDC78 | FHL1 | MTMR14 | SYNE1 |
| CFL2 | FKRP | MUSK | SYNE2 |
| CHAT | FKTN | MYBPC3 | SYT2 |
| CHKB | FLNC | MYF6 | TCAP |
| CHRNA1 | GAA | MYH2 | TIA1 |
| CHRNB1 | GARS | MYH7 | TMEM43 |
| CHRND | GFPT1 | MYO18B | TMEM5 |
| CHRNE | GMPPB | MYOT | TNNT1 |
| CHRNG | GNE | NEB | TNPO3 |
| Cntn1 | GTDC2 | PABPN1 | TPM2 |
| COL12A1 | HSPB8 | PLEC | TPM3 |
| COL6A1 | IGHMBP2 | PLEKHG5 | TRIM32 |
| COL6A2 | ISPD | POMGNT1 | TRPV4 |
| COL6A3 | ITGA7 | POMT1 | TTN |
| COLQ | KBTBD13 | POMT2 | VCP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Natera-de Benito, D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. https://doi.org/10.3390/genes11050539
Gonzalez-Quereda L, Rodriguez MJ, Diaz-Manera J, Alonso-Perez J, Gallardo E, Nascimento A, Ortez C, Natera-de Benito D, Olive M, Gonzalez-Mera L, et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes. 2020; 11(5):539. https://doi.org/10.3390/genes11050539
Chicago/Turabian StyleGonzalez-Quereda, Lidia, Maria Jose Rodriguez, Jordi Diaz-Manera, Jorge Alonso-Perez, Eduard Gallardo, Andres Nascimento, Carlos Ortez, Daniel Natera-de Benito, Montse Olive, Laura Gonzalez-Mera, and et al. 2020. "Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain" Genes 11, no. 5: 539. https://doi.org/10.3390/genes11050539
APA StyleGonzalez-Quereda, L., Rodriguez, M. J., Diaz-Manera, J., Alonso-Perez, J., Gallardo, E., Nascimento, A., Ortez, C., Natera-de Benito, D., Olive, M., Gonzalez-Mera, L., Lopez de Munain, A., Zulaica, M., Poza, J. J., Jerico, I., Torne, L., Riera, P., Milisenda, J., Sanchez, A., Garrabou, G., ... Gallano, P. (2020). Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes, 11(5), 539. https://doi.org/10.3390/genes11050539