Ecological Processes Affecting Long-Term Eukaryote and Prokaryote Biofilm Persistence in Nitrogen Removal from Sewage


Abstract
1. Introduction
2. Materials and Methods
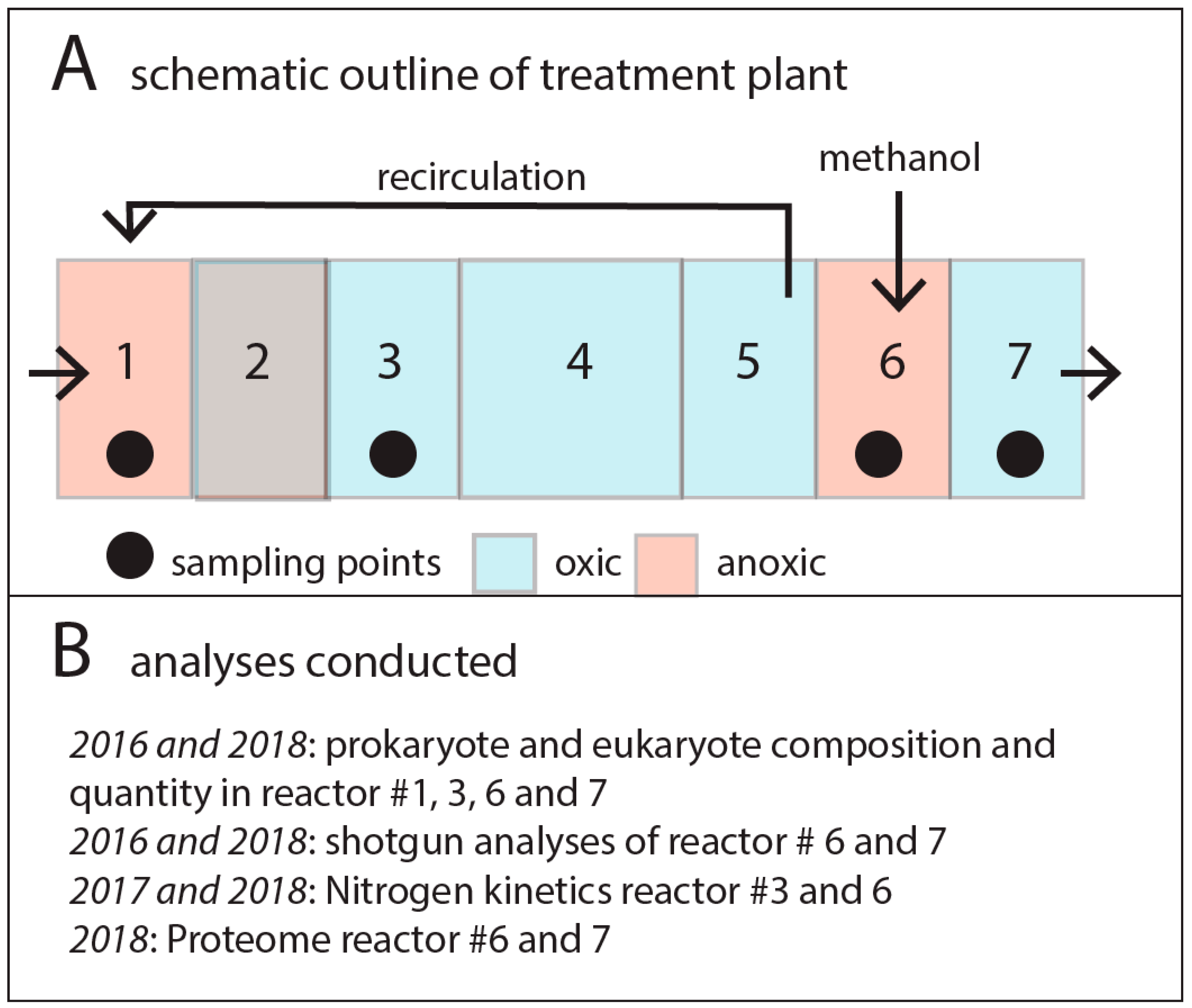
2.1. Experimental Site and Setup
2.2. DNA Extraction
2.3. Amplicon Sequencing
2.4. Whole Metagenome Sequencing
2.5. SRA Database Search
2.6. Proteome Analysis
2.7. Nitrogen Turnover in Biobeads from Reactors #3 and #6
3. Results
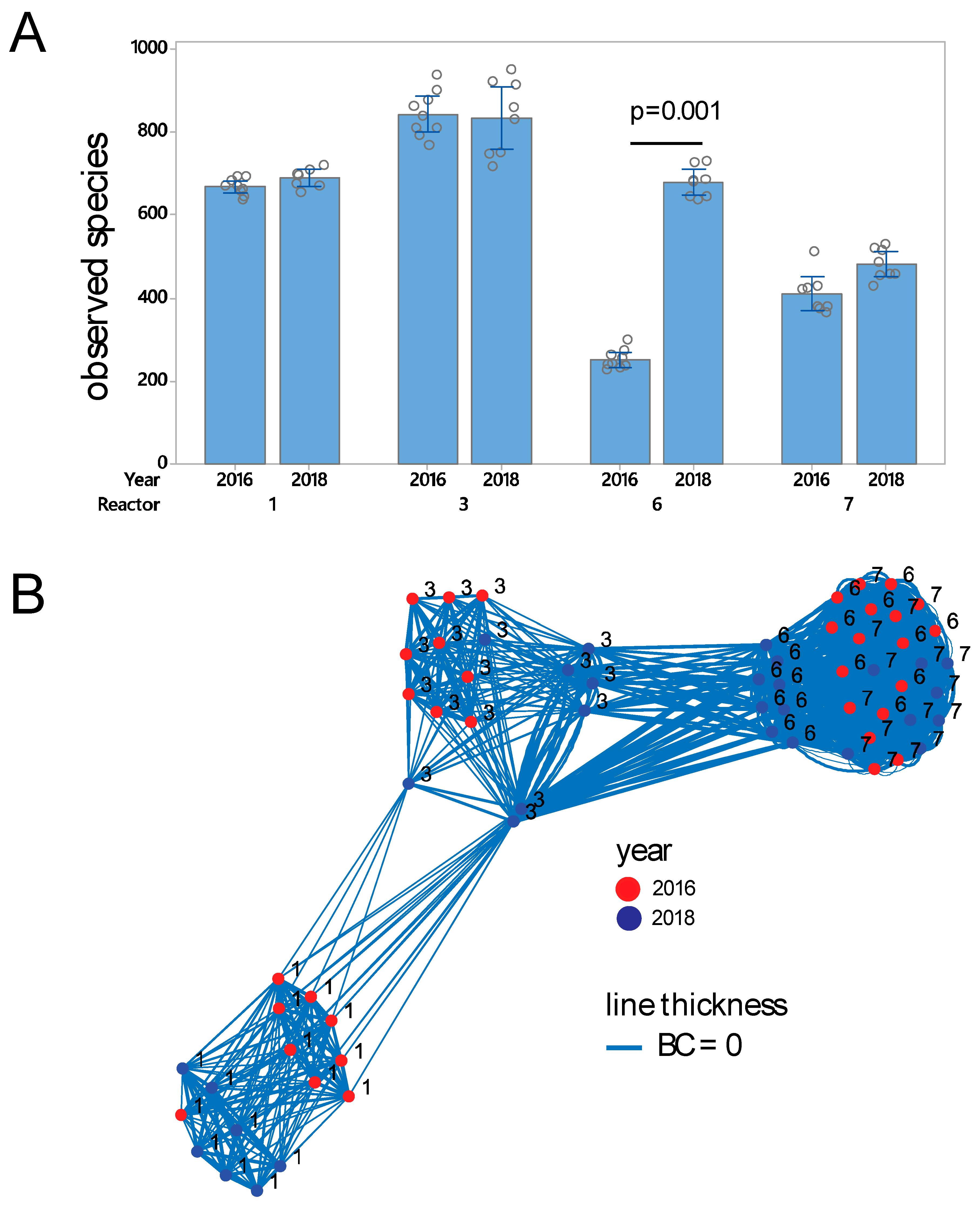
3.1. Prokaryote Composition and Diversity
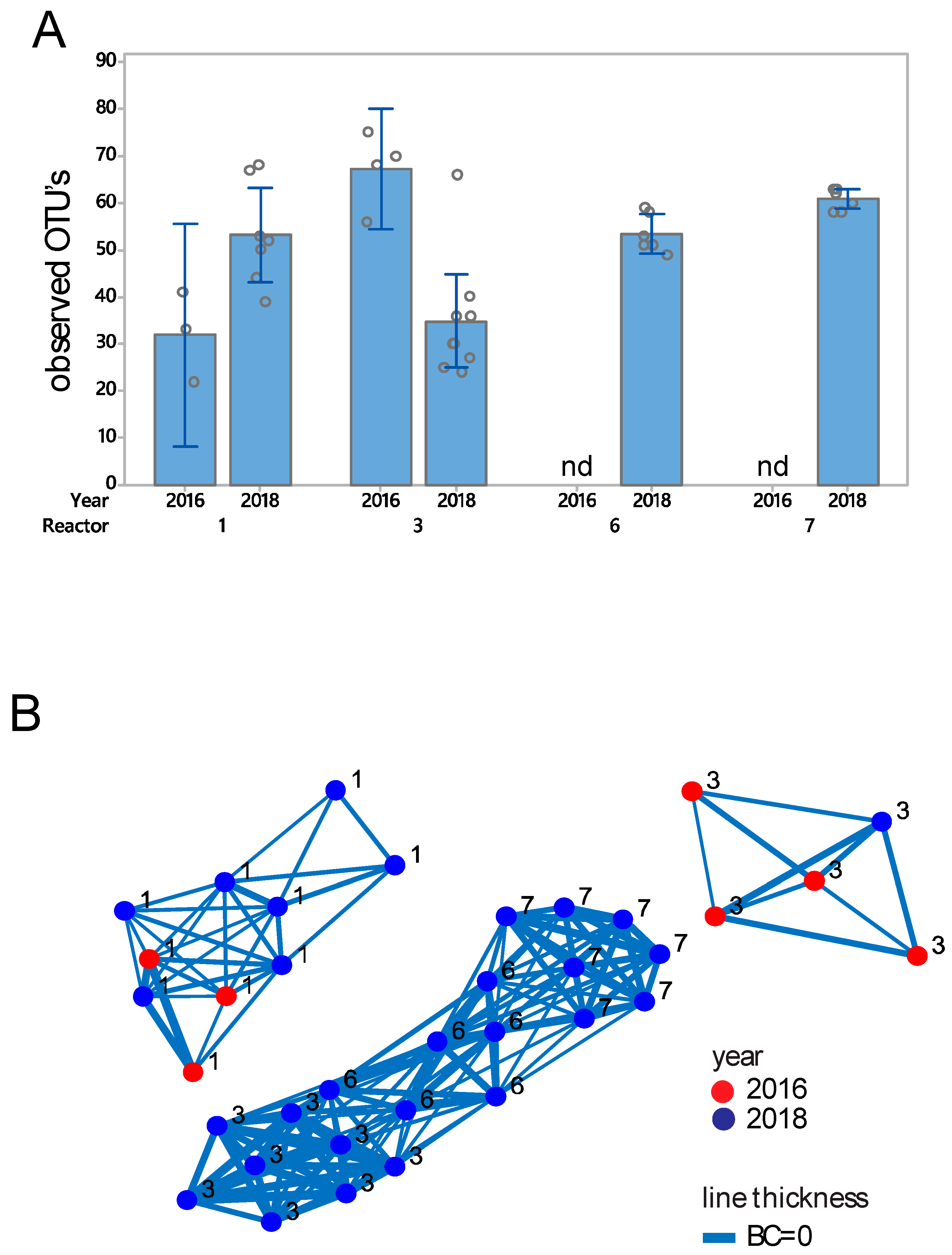
3.2. Eukaryote Microbiota Composition and Diversity
3.3. Functional and Strain Resolution Analyses in Reactors #6 and #7
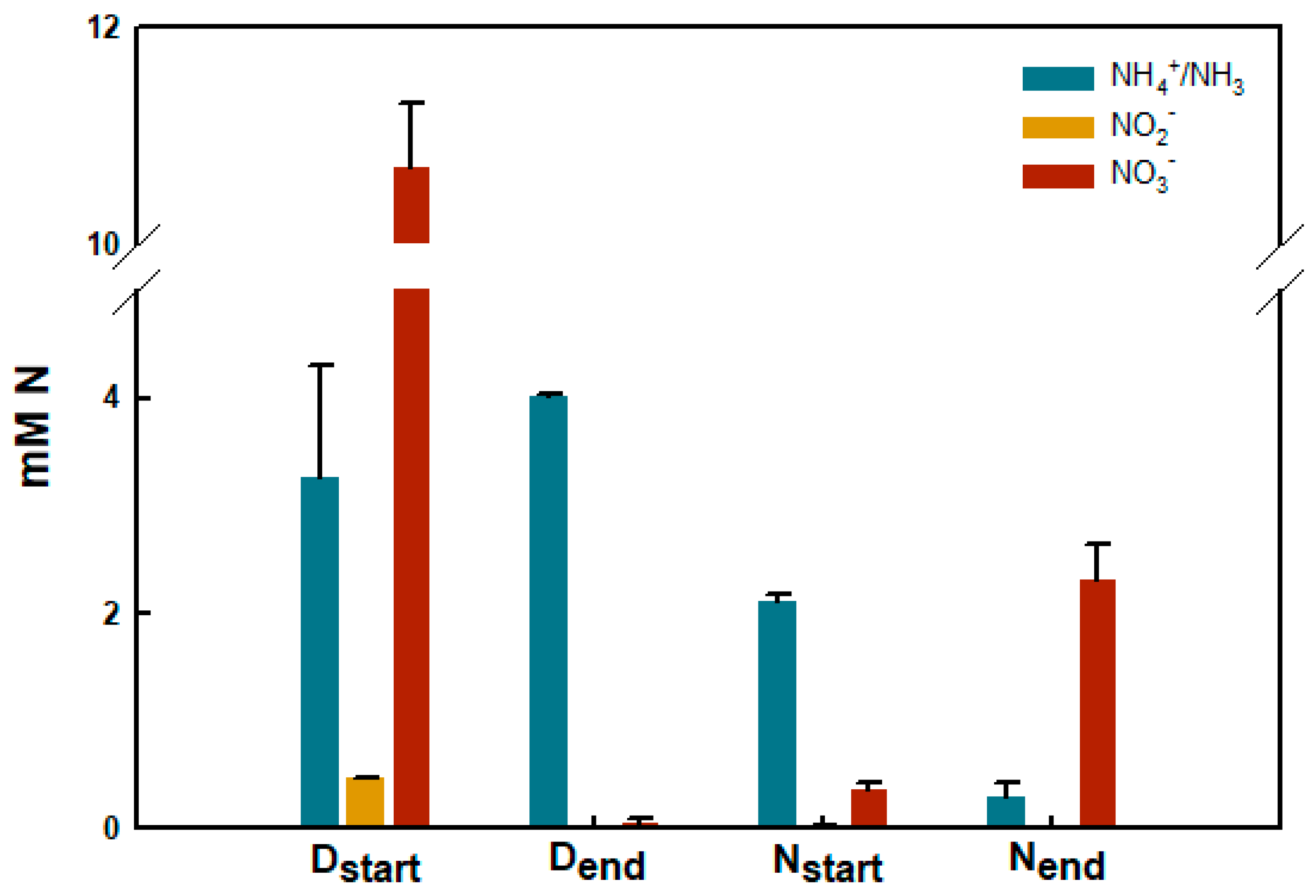
3.4. Functional Characteristics of Biobead Performance
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An emergent form of bacterial life. Nat. Rev. Microbiol. 2016, 14, 563. [Google Scholar] [CrossRef] [PubMed]
- Helness, H.; Melin, E.; Ulgenes, Y.; Jarvinen, P.; Rasmussen, V.; Odegaard, H. High-rate wastewater treatment combining a moving bed biofilm reactor and enhanced particle separation. Water Sci. Technol. 2005, 52, 117–127. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ahn, Y.-H. Sustainable nitrogen elimination biotechnologies: A review. Process Biochem. 2006, 41, 1709–1721. [Google Scholar] [CrossRef]
- Boltz, J.P.; Smets, B.F.; Rittmann, B.E.; van Loosdrecht, M.C.M.; Morgenroth, E.; Daigger, G.T. From biofilm ecology to reactors: A focused review. Water Sci. Technol. 2017, 75, 1753–1760. [Google Scholar] [CrossRef]
- Ødegaard, H.; Rusten, B.; Westrum, T. A new moving bed biofilm reactor - applications and results. Water Sci. Technol. 1994, 29, 157–165. [Google Scholar] [CrossRef]
- Torresi, E.; Fowler, S.J.; Polesel, F.; Bester, K.; Andersen, H.R.; Smets, B.F.; Plósz, B.G.; Christensson, M. Biofilm Thickness Influences Biodiversity in Nitrifying MBBRs—Implications on Micropollutant Removal. Environ. Sci. Technol. 2016, 50, 9279–9288. [Google Scholar] [CrossRef]
- Suarez, C.; Piculell, M.; Modin, O.; Langenheder, S.; Persson, F.; Hermansson, M. Thickness determines microbial community structure and function in nitrifying biofilms via deterministic assembly. Sci. Rep. 2019, 9, 5110. [Google Scholar] [CrossRef]
- Angell, I.L.; Hanssen, J.F.; Rudi, K. Prokaryote species richness is positively correlated with eukaryote abundance in wastewater treatment biofilms. Lett. Appl. Microbiol. 2017, 65, 66–72. [Google Scholar] [CrossRef]
- Rudi, K.; Goa, I.A.; Saltnes, T.; Sorensen, G.; Angell, I.L.; Eikas, S. Microbial ecological processes in MBBR biofilms for biological phosphorus removal from wastewater. Water Sci. Technol. 2019, 79, 1467–1473. [Google Scholar] [CrossRef]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Burgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.; et al. Fundamentals of microbial community resistance and resilience. Front. Microbiol. 2012, 3, 417. [Google Scholar] [CrossRef]
- De Wit, R.; Bouvier, T. ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ. Microbiol. 2006, 8, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Bell, G. Neutral macroecology. Science 2001, 293, 2413–2418. [Google Scholar] [CrossRef] [PubMed]
- Hubbell, S.P. The Unified Neutral Theory of Biodiversity and Biogeography; Princeton University Press: Princeton, NJ, USA, 2001. [Google Scholar]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002–e000017. [Google Scholar] [CrossRef] [PubMed]
- Ofiţeru, I.D.; Lunn, M.; Curtis, T.P.; Wells, G.F.; Criddle, C.S.; Francis, C.A.; Sloan, W.T. Combined niche and neutral effects in a microbial wastewater treatment community. Proc. Natl. Acad. Sci. USA 2010, 107, 15345–15350. [Google Scholar] [CrossRef] [PubMed]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and processes of microbial community assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef]
- Levi, K.; Rynge, M.; Abeysinghe, E.; Edwards, R.A. Searching the Sequence Read Archive using Jetstream and Wrangler. In Proceedings of the Practice and Experience on Advanced Research Computing; ACM: Pittsburgh, PA, USA, 2018; pp. 1–7. [Google Scholar]
- Zumft, W.G. Cell biology and molecular basis of denitrification. Microbiol. Mol. Biol. Rev. 1997, 61, 533–616. [Google Scholar] [CrossRef]
- Oswald, R.; Behrendt, T.; Ermel, M.; Wu, D.; Su, H.; Cheng, Y.; Breuninger, C.; Moravek, A.; Mougin, E.; Delon, C.; et al. HONO Emissions from Soil Bacteria as a Major Source of Atmospheric Reactive Nitrogen. Science 2013, 341, 1233–1235. [Google Scholar] [CrossRef]
- Ravishankara, A.R.; Daniel, J.S.; Portmann, R.W. Nitrous oxide (N2O): The dominant ozone-depleting substance emitted in the 21st century. Science 2009, 326, 123–125. [Google Scholar] [CrossRef]
- Yu, Y.; Lee, C.; Kim, J.; Hwang, S. Group-specific primer and probe sets to detect methanogenic communities using quantitative real-time polymerase chain reaction. Biotechnol. Bioeng. 2005, 89, 670–679. [Google Scholar] [CrossRef]
- Brate, J.; Logares, R.; Berney, C.; Ree, D.K.; Klaveness, D.; Jakobsen, K.S.; Shalchian-Tabrizi, K. Freshwater Perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA. ISME J. 2010, 4, 1144–1153. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- McIlroy, S.J.; Kirkegaard, R.H.; McIlroy, B.; Nierychlo, M.; Kristensen, J.M.; Karst, S.M.; Albertsen, M.; Nielsen, P.H. MiDAS 2.0: An ecosystem-specific taxonomy and online database for the organisms of wastewater treatment systems expanded for anaerobic digester groups. Database (Oxford) 2017, 2017. [Google Scholar] [CrossRef]
- Wattam, A.R.; Brettin, T.; Davis, J.J.; Gerdes, S.; Kenyon, R.; Machi, D.; Mao, C.; Olson, R.; Overbeek, R.; Pusch, G.D.; et al. Assembly, Annotation, and Comparative Genomics in PATRIC, the All Bacterial Bioinformatics Resource Center. Methods Mol. Biol. 2018, 1704, 79–101. [Google Scholar]
- Torres, P.J.; Edwards, R.A.; McNair, K.A. PARTIE: A partition engine to separate metagenomic and amplicon projects in the Sequence Read Archive. Bioinformatics 2017, 33, 2389–2391. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Towns, J.; Cockerill, T.; Dahan, M.; Foster, I.; Gaither, K.; Grimshaw, A.; Hazlewood, V.; Lathrop, S.; Lifka, D.; Peterson, G.D.; et al. XSEDE: Accelerating Scientific Discovery. Comput. Sci. Eng. 2014, 16, 62–74. [Google Scholar] [CrossRef]
- Zougman, A.; Selby, P.J.; Banks, R.E. Suspension trapping (STrap) sample preparation method for bottom-up proteomics analysis. Proteomics 2014, 14, 1006-0. [Google Scholar] [CrossRef]
- Molstad, L.; Dorsch, P.; Bakken, L.R. Robotized incubation system for monitoring gases (O2, NO, N2O N2) in denitrifying cultures. J. Microbiol. Methods 2007, 71, 202–211. [Google Scholar] [CrossRef]
- Freeman, K.R.; Martin, A.P.; Karki, D.; Lynch, R.C.; Mitter, M.S.; Meyer, A.F.; Longcore, J.E.; Simmons, D.R.; Schmidt, S.K. Evidence that chytrids dominate fungal communities in high-elevation soils. Proc. Natl. Acad. Sci. USA 2009, 106, 18315–18320. [Google Scholar] [CrossRef]
- Gupta, M.; Ho, D.; Santoro, D.; Torfs, E.; Doucet, J.; Vanrolleghem, P.A.; Nakhla, K. Experimental assessment and validation of quantification methods for cellulose content in municipal wastewater and sludge. Environ. Sci. Pollut. Res. Int. 2018, 25, 16743–16753. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.J.; Xi, C.; Raskin, L. Bacterial Community Structure in the Drinking Water Microbiome Is Governed by Filtration Processes. Environ. Sci. Technol. 2012, 46, 8851–8859. [Google Scholar] [CrossRef] [PubMed]
- Wells, G.F.; Wu, C.H.; Piceno, Y.M.; Eggleston, B.; Brodie, E.L.; Desantis, T.Z.; Andersen, G.L.; Hazen, T.C.; Francis, C.A.; Criddle, C.S. Microbial biogeography across a full-scale wastewater treatment plant transect: Evidence for immigration between coupled processes. Appl. Microbiol. Biotechnol. 2014, 98, 4723–4736. [Google Scholar] [CrossRef]
- Daims, H.; Lebedeva, E.V.; Pjevac, P.; Han, P.; Herbold, C.; Albertsen, M.; Nico, J.; Marton, P.; Julia, V.; Alexandr, B.; et al. Complete nitrification by Nitrospira bacteria. Nature 2015, 528, 504–509. [Google Scholar] [CrossRef]
- Mylnikova, Z.M.; Mylnikov, A.P. Structure of filose amoeba Rhogostoma minus Belar 1921 (Cryomonadida, Cercozoa) cell. Inland Water Biol. 2012, 5, 236–240. [Google Scholar] [CrossRef]
- Wildschutte, H.; Wolfe, D.M.; Tamewitz, A.; Lawrence, J.G. Protozoan predation, diversifying selection, and the evolution of antigenic diversity in Salmonella. Proc. Natl. Acad. Sci. USA 2004, 101, 10644–10649. [Google Scholar] [CrossRef] [PubMed]
- Germerodt, S.; Bohl, K.; Lück, A.; Pande, S.; Schröter, A.; Kaleta, C.; Schuster, S.; Kost, C. Pervasive Selection for Cooperative Cross-Feeding in Bacterial Communities. PLOS Comput. Biol. 2016, 12, e1004986. [Google Scholar] [CrossRef]
- Goldford, J.E.; Lu, N.; Bajić, D.; Estrela, S.; Tikhonov, M.; Sanchez-Gorostiaga, A.; Segrè, D.; Mehta, P.; Sanchez, A. Emergent simplicity in microbial community assembly. Science 2018, 361, 469–474. [Google Scholar] [CrossRef]
- Sun, Y.; Shen, D.; Zhou, X.; Shi, N.; Tian, Y. Microbial diversity and community structure of denitrifying biological filters operated with different carbon sources. Springerplus 2016, 5, 1752. [Google Scholar] [CrossRef]
- Lycus, P.; Lovise Bothun, K.; Bergaust, L.; Peele Shapleigh, J.; Reier Bakken, L.; Frostegard, A. Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J. 2017, 11, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Mao, Y.; Bergaust, L.; Bakken, L.R.; Frostegard, A. Strains in the genus Thauera exhibit remarkably different denitrification regulatory phenotypes. Environ. Microbiol. 2013, 15, 2816–2828. [Google Scholar] [PubMed]
- Saunders, A.M.; Albertsen, M.; Vollertsen, J.; Nielsen, P.H. The activated sludge ecosystem contains a core community of abundant organisms. ISME J. 2016, 10, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Leventhal, G.E.; Boix, C.; Kuechler, U.; Enke, T.N.; Sliwerska, E.; Holliger, C.; Cordero, O.X. Strain-level diversity drives alternative community types in millimetre-scale granular biofilms. Nat. Microbiol. 2018, 3, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Karst, S.M.; Gilbert, E.M.; Horn, H.; Nielsen, P.H.; Lackner, S. The role of inoculum and reactor configuration for microbial community composition and dynamics in mainstream partial nitritation anammox reactors. Microbiologyopen 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- McGill, B.J. A test of the unified neutral theory of biodiversity. Nature 2003, 422, 881–885. [Google Scholar] [CrossRef]
- O’Malley, M.A. ‘Everything is everywhere: But the environment selects’: Ubiquitous distribution and ecological determinism in microbial biogeography. Studies Hist. Philos. Sci. Part C 2008, 39, 314–325. [Google Scholar] [CrossRef]
- Bahram, M.; Kohout, P.; Anslan, S.; Harend, H.; Abarenkov, K.; Tedersoo, L. Stochastic distribution of small soil eukaryotes resulting from high dispersal and drift in a local environment. ISME J. 2016, 10, 885–896. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Name | ID 1 | Coarse Consist (%) 2 | Completeness (%) | DNA Size (bp) | r6 3 2016 | r7 3 2016 | r6 3 2018 | r7 3 2018 |
|---|---|---|---|---|---|---|---|---|
| Hyphomicrobium sp. | 113,574.7 | 74.8 | 54.94 | 2,070,006 | 0.9 | 0.7 | 0.9 | 0.4 |
| Hydrotalea flava | 714,549.25 | 88.4 | 15.74 | 274,213 | 0.2 | 0.1 | 0.1 | 0.04 |
| Methylophilus sp. | 1,112,274.5 | 82.9 | 71.41 | 2,555,358 | 6.3 | 7.1 | 1.6 | 7 |
| Methylotenera mobilis | 583,345.9 | 91.3 | 97.17 | 2,966,596 | 15.1 | 11.8 | 2.8 | 15.4 |
| Cupriavidus sp. | 367,825.5 | 86.5 | 12.52 | 511,195 | 0.5 | 0.4 | 0.4 | 0.3 |
| Methylotenera sp. | 1,506,585.5 | 87.5 | 19.59 | 811,227 | 4.4 | 3.2 | 0.9 | 4.1 |
| Flavihumibacter solisilvae | 1,349,421.6 | 83.6 | 14.17 | 434,301 | 0.1 | 0.1 | 0.2 | 0.1 |
| Polaromonas sp. | 296,591.29 | 75.7 | 49.85 | 1,455,211 | 0.9 | 0.9 | 1.6 | 0.5 |
| Rhodoferax ferrireducens | 338,969.31 | 71.5 | 44.59 | 2,352,732 | 1.5 | 1.7 | 2.5 | 0.9 |
| Luteibacter sp. | 1,798,239.6 | 75.7 | 35.08 | 1,107,355 | 0.5 | 0.4 | 0.9 | 0.3 |
| Curvibacter sp. | 1,797,748.7 | 80 | 83.62 | 2,770,618 | 2 | 2.3 | 3.5 | 1 |
| Methylobacillus sp. | 1,848,039.6 | 82.3 | 96.55 | 4,056,962 | 3.6 | 3.4 | 5.1 | 3.7 |
| Dokdonella immobilis | 578,942.18 | 80.5 | 96.55 | 8,908,583 | 1.1 | 1.4 | 4.8 | 0.5 |
| Methylotenera versatilis | 666,681.11 | 84 | 100 | 24,734,622 | 48.1 | 51.2 | 12.5 | 68.3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angell, I.L.; Bergaust, L.; Hanssen, J.F.; Aasen, E.M.; Rudi, K. Ecological Processes Affecting Long-Term Eukaryote and Prokaryote Biofilm Persistence in Nitrogen Removal from Sewage. Genes 2020, 11, 449. https://doi.org/10.3390/genes11040449
Angell IL, Bergaust L, Hanssen JF, Aasen EM, Rudi K. Ecological Processes Affecting Long-Term Eukaryote and Prokaryote Biofilm Persistence in Nitrogen Removal from Sewage. Genes. 2020; 11(4):449. https://doi.org/10.3390/genes11040449
Chicago/Turabian StyleAngell, Inga Leena, Linda Bergaust, Jon Fredrik Hanssen, Else Marie Aasen, and Knut Rudi. 2020. "Ecological Processes Affecting Long-Term Eukaryote and Prokaryote Biofilm Persistence in Nitrogen Removal from Sewage" Genes 11, no. 4: 449. https://doi.org/10.3390/genes11040449
APA StyleAngell, I. L., Bergaust, L., Hanssen, J. F., Aasen, E. M., & Rudi, K. (2020). Ecological Processes Affecting Long-Term Eukaryote and Prokaryote Biofilm Persistence in Nitrogen Removal from Sewage. Genes, 11(4), 449. https://doi.org/10.3390/genes11040449