The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers–Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Analysis
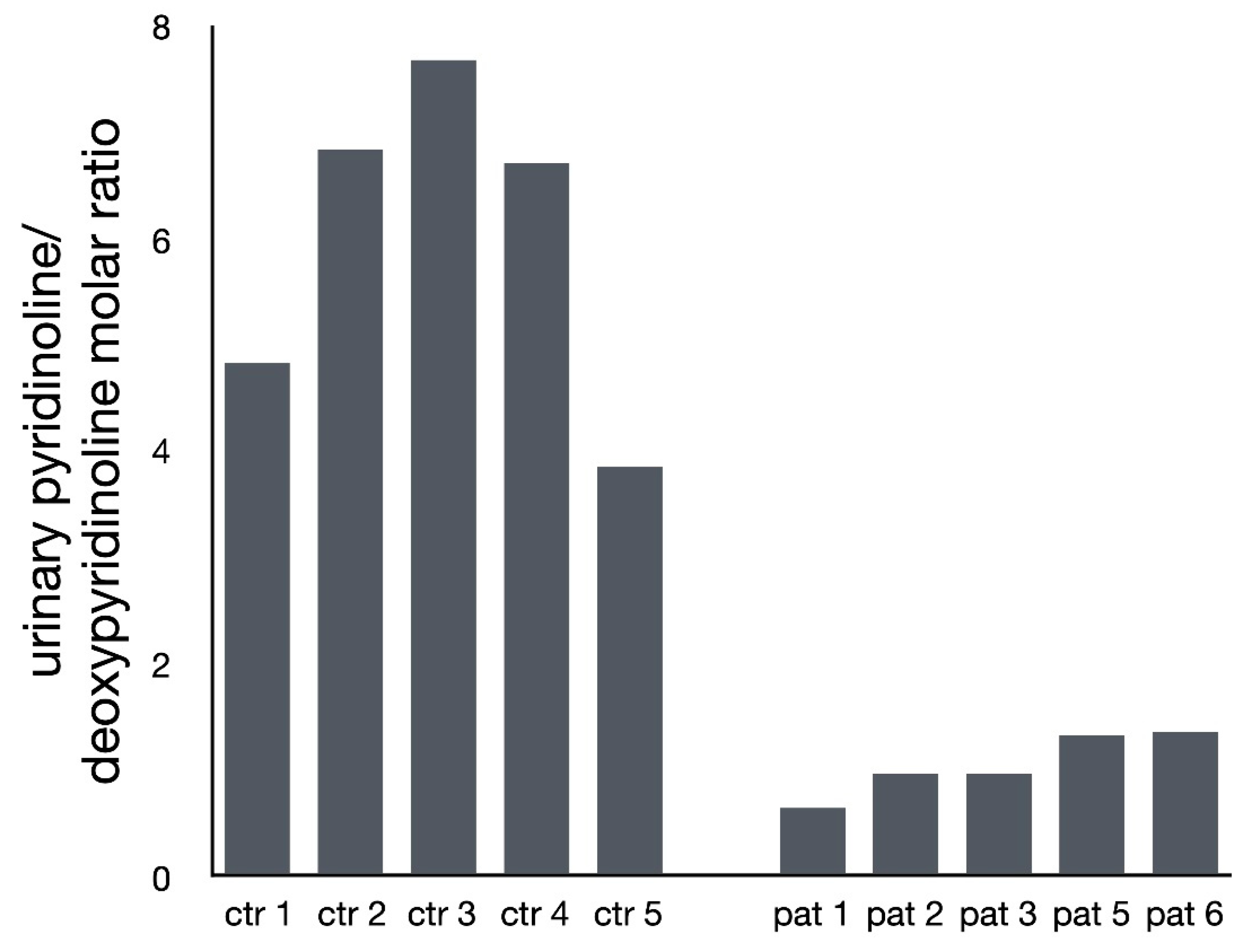
2.2. Analysis of Pyridinium Crosslink Products by HPLC
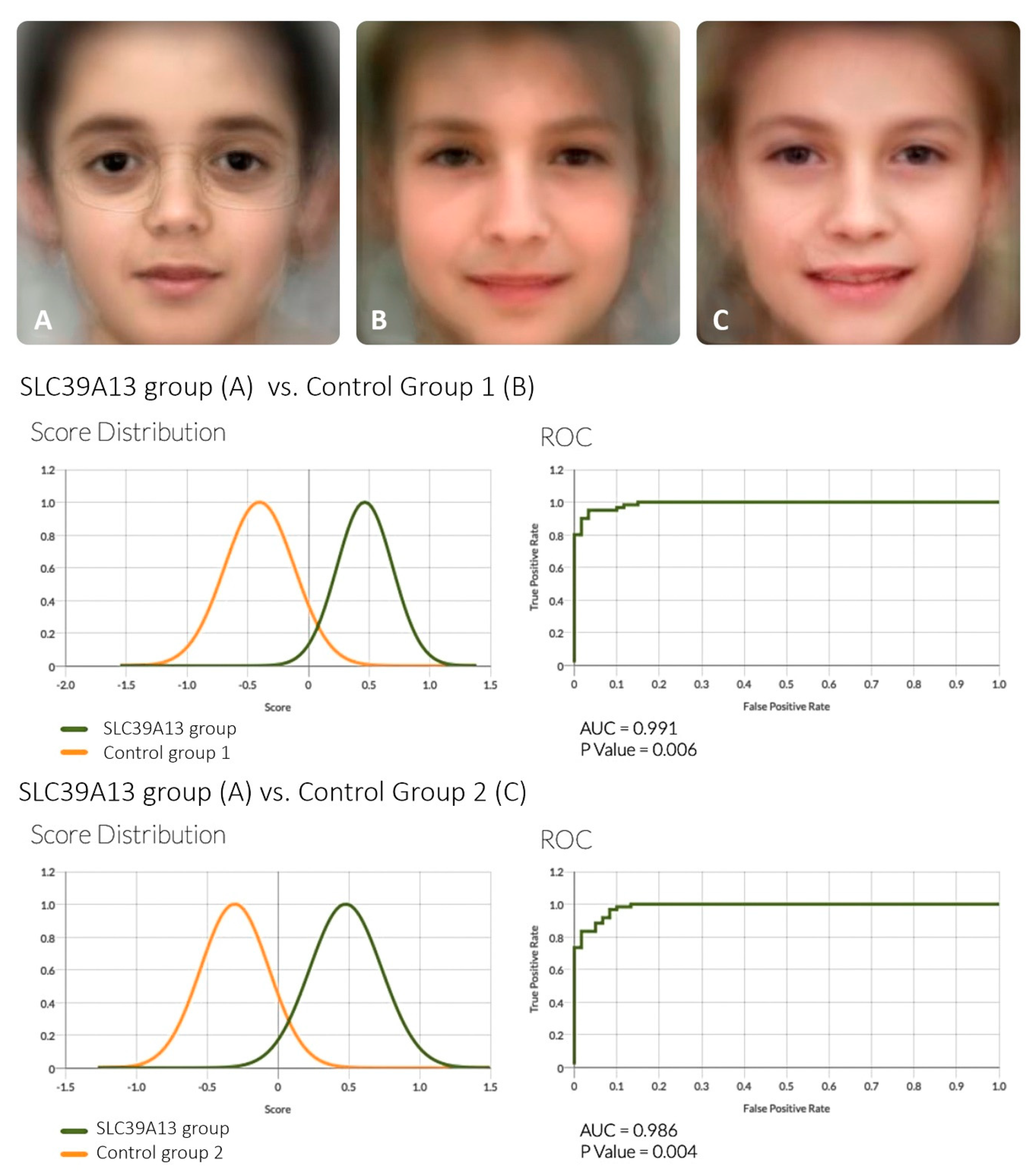
2.3. Analysis of Facial Features
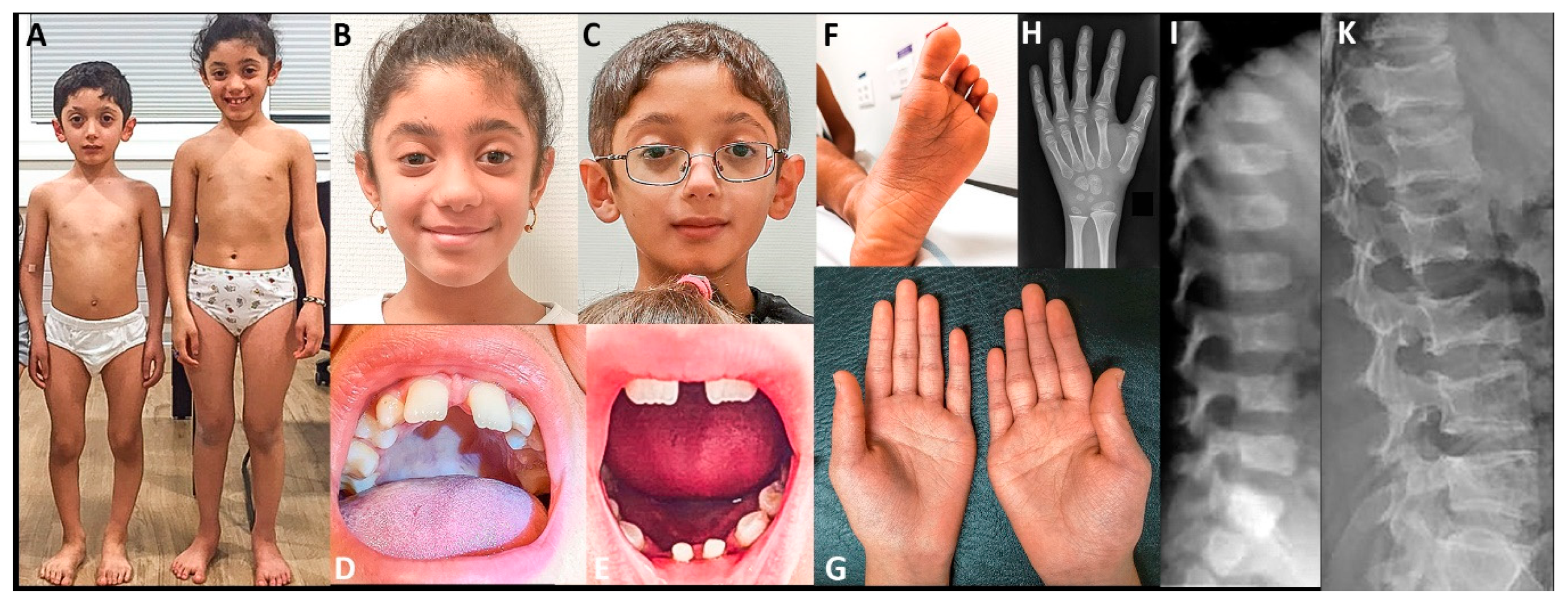
2.4. Clinical Reports
2.4.1. Individuals 1 and 2 (reported in part by Fukada et al., 2008 [5])
2.4.2. Individual 3
2.4.3. Individual 4
2.4.4. Individuals 5 and 6
3. Results
4. Discussion
4.1. Clinical Aspects of SCL39A13 Deficiency
4.2. What Are the Pathogenetic Mechanisms Leading from SLC39A13 Loss of Function to the Complex Clinical Phenotype?
4.3. Diagnosis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- The Ehlers-Danlos Syndrome. In Connective Tissue and Its Heritable Disorders; Wiley-Liss, Inc.: New York, NY, USA, 2002; pp. 431–523. [CrossRef]
- Beighton, P. Ehlers-Danlos syndrome. Ann. Rheum. Dis. 1970, 29, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Giunta, C.; Elcioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef] [PubMed]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-beta signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Mortier, G.R.; Cohn, D.H.; Cormier-Daire, V.; Hall, C.; Krakow, D.; Mundlos, S.; Nishimura, G.; Robertson, S.; Sangiorgi, L.; Savarirayan, R.; et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am. J. Med. Genet. A 2019, 179, 2393–2419. [Google Scholar] [CrossRef] [PubMed]
- Dusanic, M.; Dekomien, G.; Lucke, T.; Vorgerd, M.; Weis, J.; Epplen, J.T.; Kohler, C.; Hoffjan, S. Novel Nonsense Mutation in SLC39A13 Initially Presenting as Myopathy: Case Report and Review of the Literature. Mol. Syndromol. 2018, 9, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Gurovich, Y.; Hanani, Y.; Bar, O.; Nadav, G.; Fleischer, N.; Gelbman, D.; Basel-Salmon, L.; Krawitz, P.M.; Kamphausen, S.B.; Zenker, M.; et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat. Med. 2019, 25, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Face2Gene Research app. Available online: https://www.face2gene.com/ (accessed on 4 April 2020).
- Amudhavalli, S.M.; Hanson, R.; Angle, B.; Bontempo, K.; Gripp, K.W. Further delineation of Ayme-Gripp syndrome and use of automated facial analysis tool. Am. J. Med. Genet. A 2018, 176, 1648–1656. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Murakami, M.; Fukada, T.; Nishida, K.; Yamasaki, S.; Suzuki, T. Roles of zinc and zinc signaling in immunity: Zinc as an intracellular signaling molecule. Adv. Immunol. 2008, 97, 149–176. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pat. 1 * | Pat. 2 * | Pat. 3 | Pat. 4 | Pat. 5 | Pat. 6 | |
|---|---|---|---|---|---|---|
| SLC39A13 (NM_001128225.3) | c.G221A/c.G221A | c.G221A/c.G221A | c.483_491delCTTCCTGGC/c.483_491delCTTCCTGGC | c.793G>A/c.793G>A | c.1019delT/c.1019delT | c.1019delT/c.1019delT |
| Protein (NP_001121697.2) | p.Gly74Asp | p.Gly74Asp | p.Phe162Ala164del | p.Asp265Asn | p.Leu340ProfsTer23 | p.Leu340ProfsTer23 |
| Frequency in gnomAD ** | absent | absent | extremely low, AF = 0.000009 | absent | absent | absent |
| Predictors of pathogenicity on protein *** | pathogenic | pathogenic | pathogenic | pathogenic | pathogenic | pathogenic |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumps, C.; Campos-Xavier, B.; Hilhorst-Hofstee, Y.; Marcelis, C.; Kraenzlin, M.; Fleischer, N.; Unger, S.; Superti-Furga, A. The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers–Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases. Genes 2020, 11, 420. https://doi.org/10.3390/genes11040420
Kumps C, Campos-Xavier B, Hilhorst-Hofstee Y, Marcelis C, Kraenzlin M, Fleischer N, Unger S, Superti-Furga A. The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers–Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases. Genes. 2020; 11(4):420. https://doi.org/10.3390/genes11040420
Chicago/Turabian StyleKumps, Camille, Belinda Campos-Xavier, Yvonne Hilhorst-Hofstee, Carlo Marcelis, Marius Kraenzlin, Nicole Fleischer, Sheila Unger, and Andrea Superti-Furga. 2020. "The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers–Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases" Genes 11, no. 4: 420. https://doi.org/10.3390/genes11040420
APA StyleKumps, C., Campos-Xavier, B., Hilhorst-Hofstee, Y., Marcelis, C., Kraenzlin, M., Fleischer, N., Unger, S., & Superti-Furga, A. (2020). The Connective Tissue Disorder Associated with Recessive Variants in the SLC39A13 Zinc Transporter Gene (Spondylo-Dysplastic Ehlers–Danlos Syndrome Type 3): Insights from Four Novel Patients and Follow-Up on Two Original Cases. Genes, 11(4), 420. https://doi.org/10.3390/genes11040420