Genome-Wide Identification and Analysis of P-Type Plasma Membrane H+-ATPase Sub-Gene Family in Sunflower and the Role of HHA4 and HHA11 in the Development of Salt Stress Resistance



Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Identification of P-Type PM H+-ATPases Sub-Gene Family Members in Sunflower
2.3. Phylogenetic Analysis
2.4. Gene Structure Analysis and Conserved Motif Identification
2.5. Cis-Acting Elements Analysis of PM H+-ATPase Genes Promoter Region
2.6. Expression Pattern Analysis of PM H+-ATPase Genes in Different Tissues
2.7. Expression Profiles of PM H+-ATPase Genes under Salt Stress
2.8. Subcellular Localization Determination of HHA Proteins
2.9. Plant Transformation and Salt Tolerance Test
2.9.1. Vector Construction, Arabidopsis Transformation, and Positive Transgenic Plants Identification
2.9.2. Salt Tolerant Experiment
2.9.3. Root Length Measurement
2.9.4. Na+ Content Determination
3. Results
3.1. Identification of P-Type PM H+-ATPases Sub-Gene Family Members of Sunflower
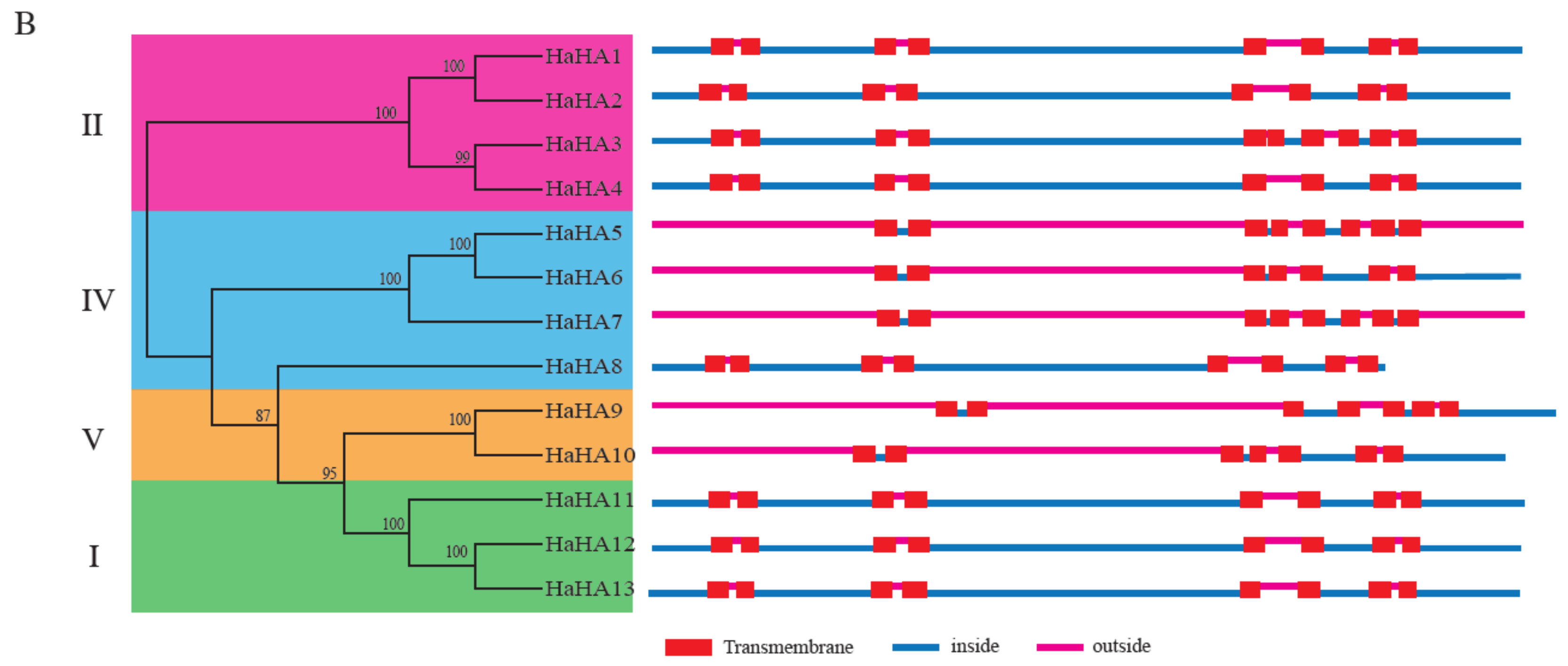
3.2. Phylogenetic Analysis
3.3. Gene Structure Analysis, Conserved Motif Identification, and Transmembrane Analysis of Sunflower PM H+-ATPases
3.4. Promoter Cis-Acting Elements Analysis
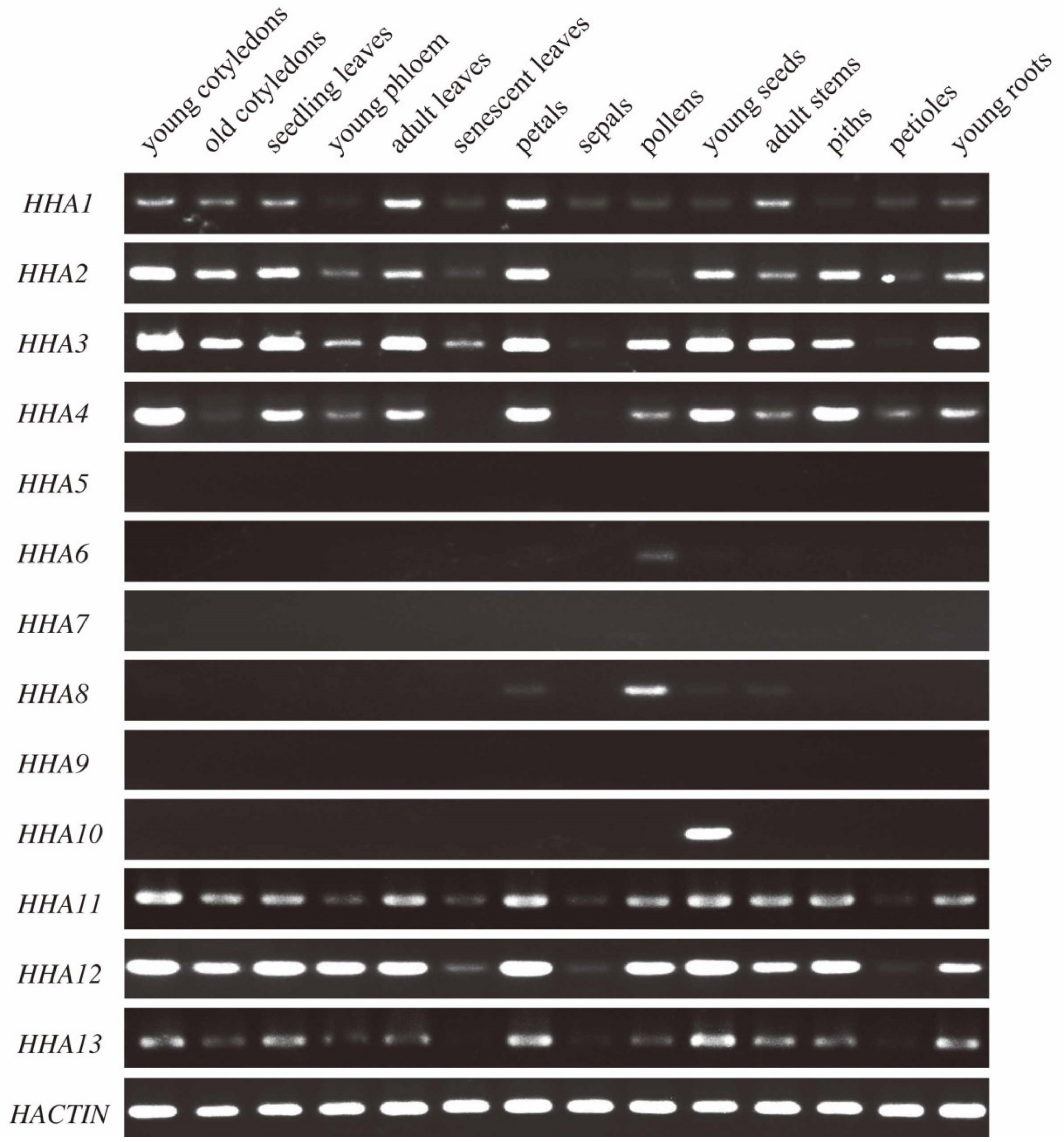
3.5. Spatiotemporal Expression Patterns of HHAs in Sunflower Tissues
3.6. Expression Patterns of HHAs in Response to Salt Stress
3.7. Subcellular Localization Analysis
3.8. Overexpression of HHA4 and HHA11 Results in Resistance to Salinity Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Axelsen, K.B.; Palmgren, M.G. Evolution of substrate specificities in the P-Type ATPase superfamily. J. Mol. Evol. 1998, 46, 84–101. [Google Scholar] [CrossRef]
- Palmgren, M.G.; Nissen, P. P-type ATPases. Annu. Rev. Biophys. 2011, 40, 243–266. [Google Scholar] [CrossRef]
- Palmgren, M.G. Plant plasma membrane H+-ATPases: Powerhouses for nutrient uptake. Annu. Rev. Plant Physiol. Plant Mol. Biol. 2001, 52, 817–845. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, P.L.; Carafoli, E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem. Sci. 1987, 12, 146–150. [Google Scholar] [CrossRef]
- Baxter, I.; Tchieu, J.; Sussman, M.R.; Boutry, M.; Palmgren, M.G.; Gribskov, M.; Harper, J.F.; Axelsen, K.B. Genomic comparison of P-Type ATPase ion pumps in Arabidopsis and rice. Plant Physiol. 2003, 132, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Lutsenko, S.; Kaplan, J.H. Organization of P-type ATPases: Significance of structural diversity. Biochemistry 1995, 34, 15607–15613. [Google Scholar] [CrossRef] [PubMed]
- MOiler, J.V.; Juul, B.; Maire, M.l. Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim. Biophys. Acta 1996, 1286, 1–51. [Google Scholar] [CrossRef]
- Bublitz, M.; Poulsen, H.; Morth, J.P.; Nissen, P. In and out of the cation pumps: P-type ATPase structure revisited. Curr. Opin. Struct. Biol. 2010, 20, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Michelet, B.; Boutry, M. The plasma membrane H+-ATPase. A highly regulated enzyme with multiple physiological functions. Plant Physiol. 1995, 108, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.B.; Shaw, B.P. Salt-inducible isoform of plasma membrane H+ATPase gene in rice remains constitutively expressed in natural halophyte, Suaeda maritima. J. Plant Physiol. 2009, 166, 1077–1089. [Google Scholar] [CrossRef] [PubMed]
- Janicka-Russak, M.; Kłobus, G. Modification of plasma membrane and vacuolar H+-ATPases in response to NaCl and ABA. J. Plant Physiol. 2007, 164, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Janicka-Russak, M.; Kabała, K. The role of plasma membrane H+-ATPase in salinity stress of plants. Prog. Bot. 2015, 76, 77–92. [Google Scholar]
- Bose, J.; Rodrigo-Moreno, A.; Lai, D.; Xie, Y.; Shen, W.; Shabala, S. Rapid regulation of the plasma membrane H+-ATPase activity is essential to salinity tolerance in two halophyte species, Atriplex lentiformis and Chenopodium quinoa. Ann. Bot. 2015, 115, 481–494. [Google Scholar] [CrossRef]
- Kalampanayil, B.D.; Wimmers, L.E. Identification and characterization of a salt-stress-induced plasma membrane H+-ATPase in tomato. Plant Cell Environ. 2001, 24, 999–1005. [Google Scholar] [CrossRef]
- Arango, M.; Gevaudant, F.; Oufattole, M.; Boutry, M. The plasma membrane proton pump ATPase: The significance of gene subfamilies. Planta 2003, 216, 355–365. [Google Scholar] [CrossRef]
- Binzel, M.L. NaCl-induced accumulation of tonoplast and plasma membrane H+-ATPase message in tomato. Physiol. Plant. 1995, 94, 722–728. [Google Scholar] [CrossRef]
- Gevaudant, F.; Duby, G.; von Stedingk, E.; Zhao, R.; Morsomme, P.; Boutry, M. Expression of a constitutively activated plasma membrane H+-ATPase alters plant development and increases salt tolerance. Plant Physiol. 2007, 144, 1763–1776. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Sun, J.; Ding, M.; Deng, S.; Hou, P.; Ma, X.; Zhang, Y.; Wang, F.; Sa, G.; et al. Overexpression of PeHA1 enhances hydrogen peroxide signaling in salt-stressed Arabidopsis. Plant Physiol. Biochem. 2013, 71, 37–48. [Google Scholar] [CrossRef]
- Kasamo, K. Regulation of plasma membrane H+-ATPase activity by the membrane environment. J. Plant Res. 2003, 116, 517–523. [Google Scholar] [CrossRef]
- Takahashi, M.; Kondou, Y.; Toyoshima, C. Interdomain communication in calcium pump as revealed in the crystal structures with transmembrane inhibitors. Proc. Natl. Acad. Sci. USA 2007, 104, 5800–5805. [Google Scholar] [CrossRef] [PubMed]
- Maudoux, O.; Batoko, H.; Oecking, C.; Gevaert, K.; Vandekerckhove, J.; Boutry, M.; Morsomme, P. A plant plasma membrane H+-ATPase expressed in yeast is activated by phosphorylation at its penultimate residue and binding of 14-3-3 regulatory proteins in the absence of fusicoccin. J. Biol. Chem. 2000, 275, 17762–17770. [Google Scholar] [CrossRef] [PubMed]
- Piette, A.-S.; Waelkens, E.; Boutry, M.; Duby, G. A phosphorylation in the c-terminal auto-inhibitory domain of the plant plasma membrane H+-ATPase activates the enzyme with no requirement for regulatory 14-3-3 proteins. J. Biol. Chem. 2011, 286, 18474–18482. [Google Scholar] [CrossRef] [PubMed]
- Morandini, P.; Valera, M.; Albumi, C.; Bonza, M.C.; Giacometti, S.; Ravera, G.; Murgia, I.; Soave, C.; Michelis, M.I.D. A novel interaction partner for the C-terminus of Arabidopsis thaliana plasma membrane H+-ATPase (AHA1 isoform): Site and mechanism of action on H+-ATPase activity differ from those of 14-3-3 proteins. Plant J. 2002, 31, 487–497. [Google Scholar] [CrossRef]
- García, M.N.M.; Muro, M.C.; Mazzocchi, L.C.; País, S.M.; Stritzler, M.; Schlesinger, M.; Capiati, D.A. The protein phosphatase 2A catalytic subunit StPP2Ac2b acts as a positive regulator of tuberization induction in Solanum tuberosum L. Plant Mol. Biol. 2016, 93, 227–245. [Google Scholar] [CrossRef]
- Stritzler, M.; García, M.N.M.; Schlesinger, M.; Cortelezzi, J.I.; Capiati, D.A. The plasma membrane H+-ATPase gene family in Solanum tuberosum L. role of PHA1 in tuberization. J. Exp. Bot. 2017, 68, 4821–4837. [Google Scholar] [CrossRef]
- Santi, S.; Locci, G.; Monte, R.; Pinton, R.; Varanini, Z. Induction of nitrate uptake in maize roots: Expression of a putative high-affinity nitrate transporter and plasma membrane H+-ATPase isoforms. J. Exp. Bot. 2003, 54, 1851–1864. [Google Scholar] [CrossRef]
- Wdowikowska, A.; Klobus, G. The plasma membrane proton pump gene family in cucumber. Acta Physiol. Plant. 2016, 38, 135. [Google Scholar] [CrossRef]
- Badouin, H.; Gouzy, J.; Grassa, C.J.; Murat, F.; Staton, S.E.; Cottret, L.; Lelandais-Brière, C.; Owens, G.L.; Carrère, S.; Mayjonade, B.; et al. The sunflower genome provides insights into oil metabolism, flowering and Asterid evolution. Nature 2017, 546, 148–152. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, M.; Shi, D.; Zhou, G.; Niu, T.; Hahn, M.G.; O’Neill, M.A.; Kong, Y. DGE-seq analysis of MUR3-related Arabidopsis mutants provides insight into how dysfunctional xyloglucan affects cell elongation. Plant Sci. 2017, 258, 156–169. [Google Scholar] [CrossRef]
- Wang, M.; Xu, Z.; Ding, A.; Kong, Y. Genome-wide identification and expression profiling analysis of the xyloglucan endotransglucosylase/hydrolase gene family in tobacco (Nicotiana tabacum L.). Genes 2018, 9, 273. [Google Scholar] [CrossRef] [PubMed]
- Chou, K.; Shen, H. Plant-mPLoc: A top-down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 2010, 5, e11335. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Jin, J.; Guo, A.; Zhang, H.; Luo, J.; Gao, G. GSDS 2.0: An upgraded gene feature visualization server. Bioinformatics 2015, 31, 1296–1297. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Williams, N.; Misleh, C.; Li, W.W. MEME: Discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006, 34, W369–W373. [Google Scholar] [CrossRef]
- Hu, D.; Xie, Q.; Liu, Q.; Zuo, T.; Zhang, H.; Zhang, Y.; Lian, X.; Zhu, L. Genome-wide distribution, expression and function analysis of the U-Box gene family in Brassica oleracea L. Genes 2019, 10, 1000. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Batoko, H.; Zheng, H.-Q.; Hawes, C.; Moore, I. A Rab1 GTPase is required for transport between the endoplasmic reticulum and golgi apparatus and for normal golgi movement in plants. Plant Cell 2000, 12, 2201–2217. [Google Scholar] [CrossRef]
- Clough, S.J.; Bent, A.F. Floral dip: A simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998, 16, 735–743. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magalhaes, P.J.; Ram, S.J. Image processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Shao, T.; Gu, X.; Zhu, T.; Pan, X.; Zhu, Y.; Long, X.; Shao, H.; Liu, M.; Rengel, Z. Industrial crop Jerusalem artichoke restored coastal saline soil quality by reducing salt and increasing diversity of bacterial community. Appl. Soil Ecol. 2019, 138, 195–206. [Google Scholar] [CrossRef]
- Sondergaard, T.E.; Schulz, A.; Palmgren, M.G. Energization of transport processes in plants. roles of the plasma membrane H+-ATPase. Plant Physiol. 2004, 136, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Morsomme, P.; Boutry, M. The plant plasma membrane H+-ATPase: Structure, function and regulation. Biochim. Biophys. Acta 2000, 1465, 1–16. [Google Scholar] [CrossRef]
- DeWitt, N.D.; Hong, B.; Sussman, M.R.; Harper, J.F. Targeting of two Arabidopsis H+-ATPase isoforms to the plasma membrane. Plant Physiol. Biochem. 1996, 112, 833–844. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ewing, N.N.; Bennett, A.B. Assessment of the number and expression of P-type H+-ATPase genes in tomato. Plant Physiol. 1994, 106, 547–557. [Google Scholar] [CrossRef]
- Zhao, R.; Dielen, V.; Kinet, J.-M.; Boutry, M. Cosuppression of a plasma membrane H+-ATPase isoform impairs sucrose translocation, stomatal opening, plant growth, and male fertility. Plant Cell 2000, 12, 535–546. [Google Scholar] [PubMed]
- Frías, I.; Caldeira, M.T.; Pérez-Castiñeira, J.R.; Navarro-Aviñó, J.P.; Culiañez-Maciá, F.A.; Kuppinger, O.; Stransky, H.; Pagés, M.; Hager, A.; Serrano, R. A major isoform of the maize plasma membrane H+-ATPase: Characterization and induction by auxin in coleoptiles. Plant Cell 1996, 8, 1533–1544. [Google Scholar]
- Zimmermann, P.; Hirsch-Hoffmann, M.; Hennig, L.; Gruissem, W. GENEVESTIGATOR. Arabidopsis microarray database and analysis toolbox. Plant Physiol. 2004, 136, 2621–2632. [Google Scholar] [CrossRef]
- Liang, W.; Ma, X.; Wan, P.; Liu, L. Plant salt-tolerance mechanism: A review. Biochem. Biophys. Res. Commun. 2018, 495, 286–291. [Google Scholar] [CrossRef]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef]
- Keisham, M.; Mukherjee, S.; Bhatla, S.C. Mechanisms of sodium transport in plants-progresses and challenges. Int. J. Mol. Sci. 2018, 19, 647. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Narasimhan, M.L.; Salzman, R.A.; Bressan, R.A.; Hasegawa, P.M. NaCl regulation of plasma membrane H+-ATPase gene expression in a glycophyte and a halophyte. Plant Physiol. 1993, 103, 713–718. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Gene Name | Accession NO. | Chr | Genomic Location | Orthologous in Arabidopsis | Identity with Arabidopsis (%) | Protein length | Molecular Weight MW (Da) | PI | Subcellular Location | Intron |
|---|---|---|---|---|---|---|---|---|---|---|---|
| II | HHA1 | HanXRQChr12g0358351 | C12 | 13551328 - 13560479 | AT2G18960 | 85.84 | 954 | 104986.84 | 7 | Plasma membrane | 13 |
| II | HHA2 | HanXRQChr10g0308521 | C10 | 205602015 - 205610779 | AT5G57350 | 85.67 | 942 | 103947.71 | 7 | Plasma membrane | 12 |
| II | HHA3 | HanXRQChr05g0142281 | C05 | 103008186 - 103001095 | AT5G57350 | 85.77 | 954 | 105039.85 | 6 | Plasma membrane | 14 |
| II | HHA4 | HanXRQChr06g0179401 | C06 | 55584733 - 55575412 | AT5G57350 | 85.35 | 954 | 105292.35 | 7 | Plasma membrane | 15 |
| IV | HHA5 | HanXRQChr02g0034901 | C02 | 19396893 - 19388276 | AT1G80660 | 83.95 | 953 | 105009.63 | 6 | Plasma membrane | 13 |
| IV | HHA6 | HanXRQChr04g0098641 | C04 | 17528781 - 17534812 | AT1G80660 | 83.53 | 953 | 104886.54 | 6 | Plasma membrane | 12 |
| IV | HHA7 | HanXRQChr16g0500091 | C16 | 8389529 - 8397496 | AT1G80660 | 83.87 | 955 | 105464.42 | 6 | Plasma membrane | 13 |
| IV | HHA8 | HanXRQChr10g0314521 | C10 | 228958293 - 228963082 | AT3G42640 | 78.80 | 851 | 93419.13 | 5 | Plasma membrane | 6 |
| V | HHA9 | HanXRQChr13g0423171 | C13 | 186222209 - 186239644 | AT5G62670 | 79.48 | 1050 | 115861.86 | 8 | Plasma membrane | 19 |
| V | HHA10 | HanXRQChr16g0509551 | C16 | 64253539 - 64258063 | AT3G47950 | 79.32 | 937 | 102808.16 | 7 | Plasma membrane | 14 |
| I | HHA11 | HanXRQChr17g0547551 | C17 | 51582188 - 51588359 | AT3G47950 | 87.12 | 959 | 105493.54 | 6 | Plasma membrane | 20 |
| I | HHA12 | HanXRQChr05g0157971 | C05 | 199927699 - 199933355 | AT3G47950 | 92.36 | 957 | 105363.3 | 6 | Plasma membrane | 20 |
| I | HHA13 | HanXRQChr08g0235741 | C08 | 141687703 - 141693862 | AT3G47950 | 91.83 | 957 | 105333.27 | 6 | Plasma membrane | 20 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, Z.; Marowa, P.; Liu, H.; Du, H.; Zhang, C.; Li, Y. Genome-Wide Identification and Analysis of P-Type Plasma Membrane H+-ATPase Sub-Gene Family in Sunflower and the Role of HHA4 and HHA11 in the Development of Salt Stress Resistance. Genes 2020, 11, 361. https://doi.org/10.3390/genes11040361
Xu Z, Marowa P, Liu H, Du H, Zhang C, Li Y. Genome-Wide Identification and Analysis of P-Type Plasma Membrane H+-ATPase Sub-Gene Family in Sunflower and the Role of HHA4 and HHA11 in the Development of Salt Stress Resistance. Genes. 2020; 11(4):361. https://doi.org/10.3390/genes11040361
Chicago/Turabian StyleXu, Zongchang, Prince Marowa, Han Liu, Haina Du, Chengsheng Zhang, and Yiqiang Li. 2020. "Genome-Wide Identification and Analysis of P-Type Plasma Membrane H+-ATPase Sub-Gene Family in Sunflower and the Role of HHA4 and HHA11 in the Development of Salt Stress Resistance" Genes 11, no. 4: 361. https://doi.org/10.3390/genes11040361
APA StyleXu, Z., Marowa, P., Liu, H., Du, H., Zhang, C., & Li, Y. (2020). Genome-Wide Identification and Analysis of P-Type Plasma Membrane H+-ATPase Sub-Gene Family in Sunflower and the Role of HHA4 and HHA11 in the Development of Salt Stress Resistance. Genes, 11(4), 361. https://doi.org/10.3390/genes11040361