Functional Annotation and Curation of Hypothetical Proteins Present in A Newly Emerged Serotype 1c of Shigella flexneri: Emphasis on Selecting Targets for Virulence and Vaccine Design Studies

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
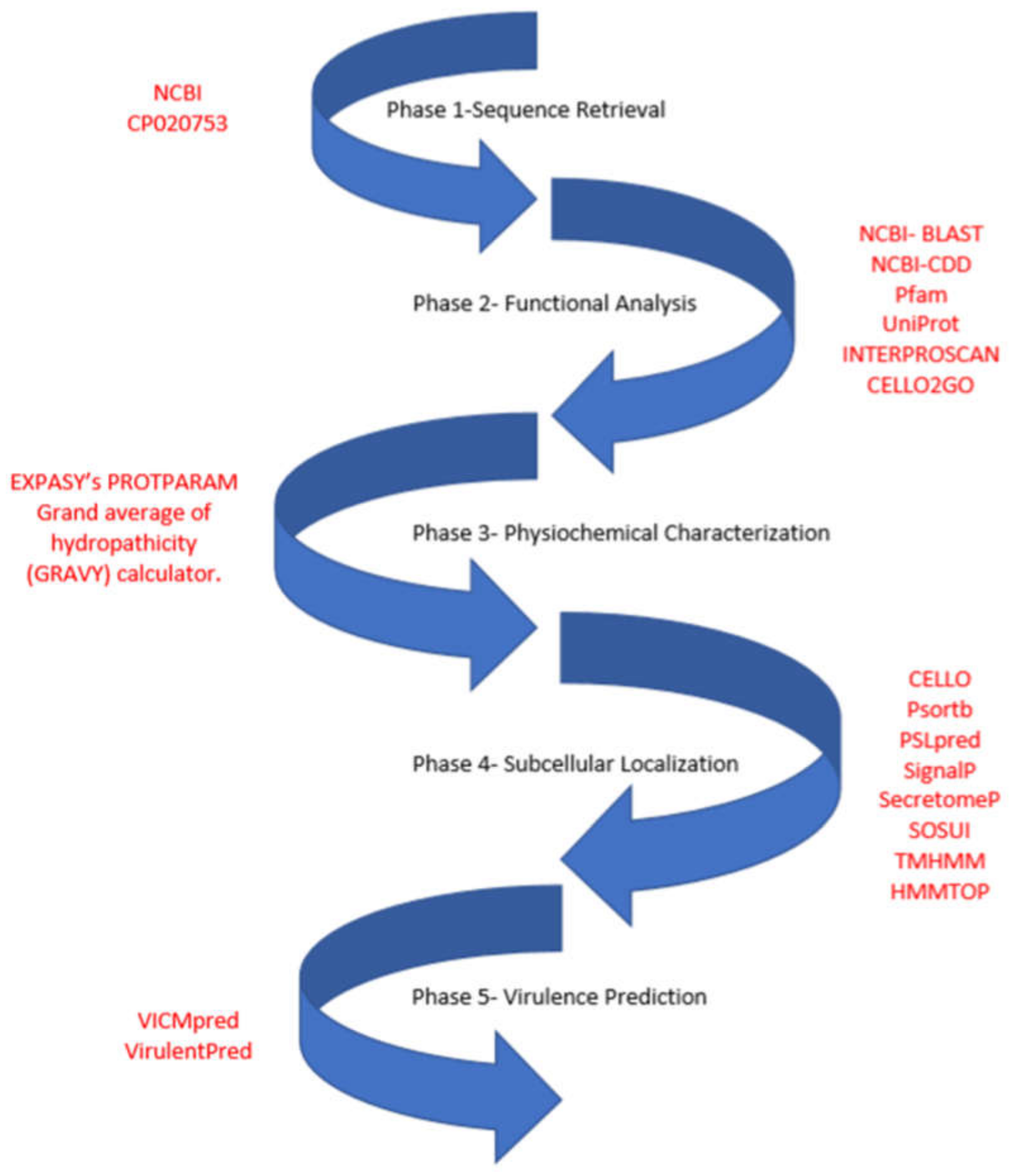
2. Materials and Methods
2.1. Functional Assignment and Domain Analysis
2.2. Physiochemical Characterization
2.3. Subcellular Localization Analysis
2.4. Virulence Factor Prediction
3. Results and Discussion
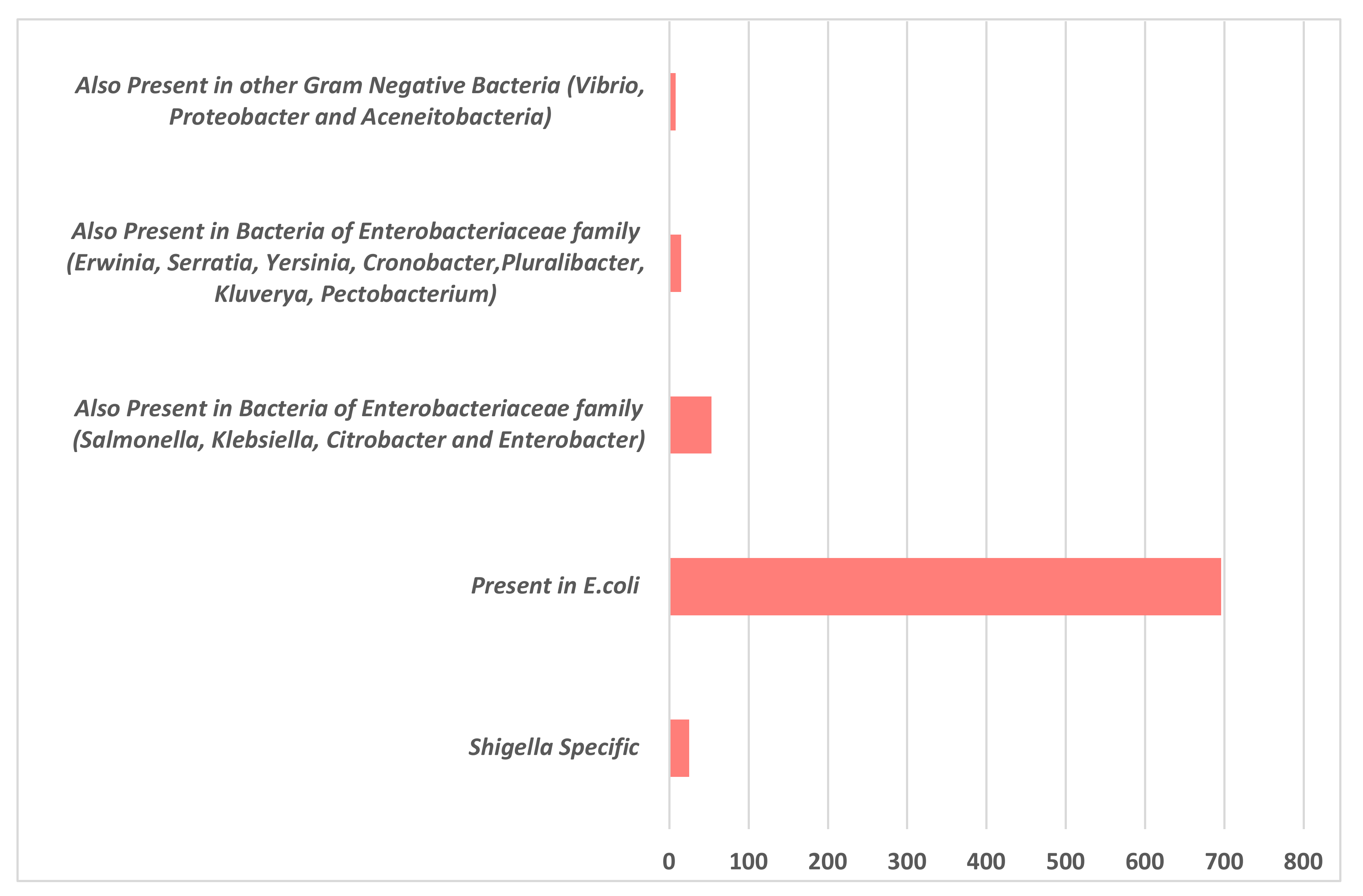
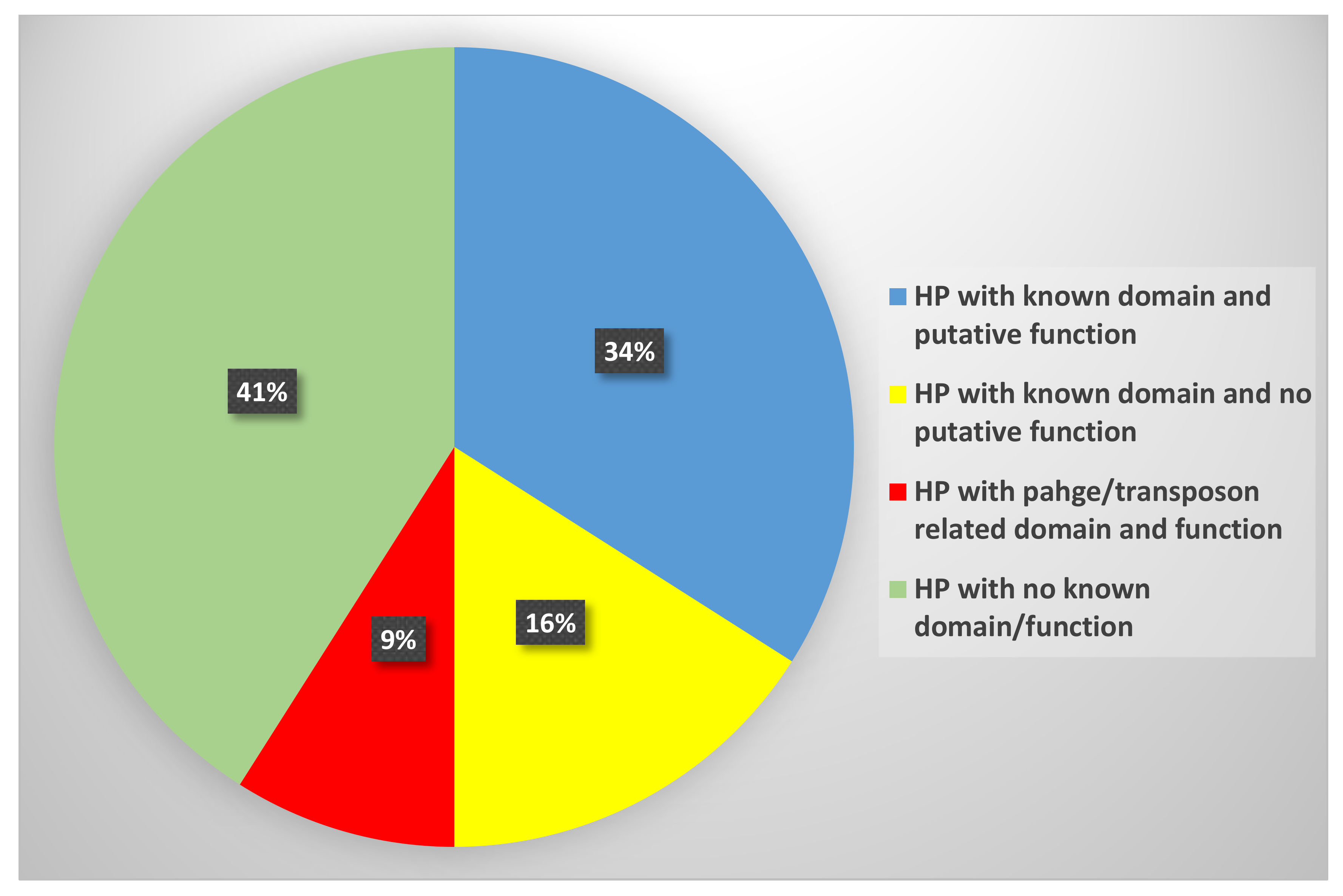
3.1. Sequence Analysis and Functional Annotation
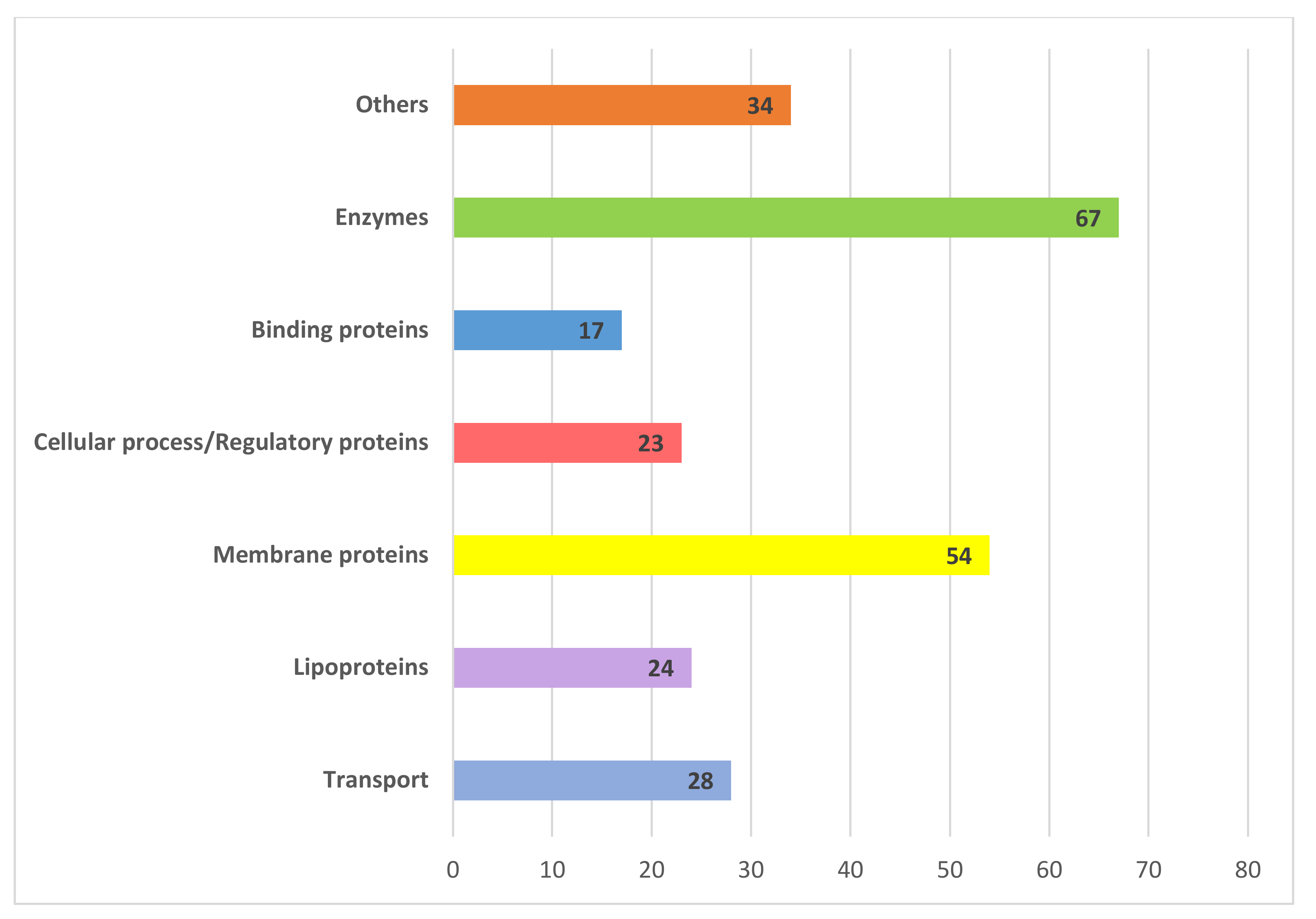
3.2. Transport Proteins
3.3. Binding Proteins
3.4. Lipoproteins
3.5. Membrane Proteins
3.6. Enzymes
3.7. Cellular Process/Regulatory Proteins
3.8. Physiochemical and Subcellular Localization Analysis
3.9. Virulence Factor Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trofa, A.F.; Ueno-Olsen, H.; Oiwa, R.; Yoshikawa, M. Dr. Kiyoshi Shiga: Discoverer of the Dysentery Bacillus. Clin. Infect. Dis. 1999, 29, 1303–1306. [Google Scholar] [CrossRef] [PubMed]
- Troeger, C.; Forouzanfar, M.; Rao, P.C.; Khalil, I.; Brown, A.; Robert, C.; Fullman, N.; Thompson, R.L.; Abajobir, A.; Ahmed, M.; et al. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef]
- Anderson, M.; Sansonetti, P.J.; Marteyn, B. Shigella Diversity and Changing Landscape: Insights for the Twenty-First Century. Front. Microbiol. 2016, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, G.; Hilbi, H. Molecular Pathogenesis of Shigella spp.: Controlling Host Cell Signaling, Invasion, and Death by Type III Secretion. Clin. Microbiol. Rev. 2008, 21, 134–156. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Sansonetti, P.J. Shigella Pathogenesis: New Insights through Advanced Methodologies. Microbiol. Spectr. 2019, 7, 15–39. [Google Scholar]
- Mathan, M.M.; Mathan, V.I. Ultrastructural pathology of the rectal mucosa in Shigella dysentery. Am. J. Pathol. 1986, 123, 25–38. [Google Scholar]
- Sethuvel, D.M.; Ragupathi, N.D.; Anandan, S.; Veeraraghavan, B. Update on: Shigella new serogroups/serotypes and their antimicrobial resistance. Lett. Appl. Microbiol. 2016, 64, 8–18. [Google Scholar] [CrossRef]
- Keusch, G.T. Shigella infections. Clin. Gastroenterol. 1979, 8, 645–662. [Google Scholar]
- Kotloff, K.L.; Riddle, M.S.; A Platts-Mills, J.; Pavlinac, P.; Zaidi, A.K.M. Shigellosis. Lancet 2018, 391, 801–812. [Google Scholar] [CrossRef]
- Hosangadi, D.; Smith, P.G.; Giersing, B.K. Considerations for using ETEC and Shigella disease burden estimates to guide vaccine development strategy. Vaccine 2019, 37, 7372–7380. [Google Scholar] [CrossRef]
- Taneja, N.; Mewara, A. Shigellosis: Epidemiology in India. Indian J. Med Res. 2016, 143, 565–576. [Google Scholar] [CrossRef]
- Parajuli, P.; Adamski, M.; Verma, N. Bacteriophages are the major drivers of Shigella flexneri serotype 1c genome plasticity: A complete genome analysis. BMC Genom. 2017, 18, 722. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Carlin, N.; Majcher, M.; Tabor, H.; Ng, L.-K.; Widmalm, G. Structural elucidation of the O-antigen of the Shigella flexneri provisional serotype 88-893: Structural and serological similarities with S. flexneri provisional serotype Y394 (1c). Carbohydr. Res. 2011, 346, 872–876. [Google Scholar] [CrossRef] [PubMed]
- Wehler, T.; Carlin, N.I.A. Structural and immunochemical studies of the lipopolysaccharide from a new provisional serotype of Shigella flexneri. JBIC J. Boil. Inorg. Chem. 1988, 176, 471–476. [Google Scholar] [CrossRef] [PubMed]
- El-Gendy, A.; El-Ghorab, N.; Lane, E.M.; Abu Elyazeed, R.; Carlin, N.I.A.; Mitry, M.M.; Kay, B.A.; Savarino, S.J.; Peruski, L.F. Identification of Shigella flexneriSubserotype 1c in Rural Egypt. J. Clin. Microbiol. 1999, 37, 873–874. [Google Scholar] [CrossRef]
- Stagg, R.M.; Tang, S.-S.; Carlin, N.I.A.; Talukder, K.A.; Cam, P.D.; Verma, N. A Novel Glucosyltransferase Involved in O-Antigen Modification of Shigella flexneri Serotype 1c. J. Bacteriol. 2009, 191, 6612–6617. [Google Scholar] [CrossRef]
- Von Seidlein, L.; Kim, D.R.; Ali, M.; Lee, H.; Wang, X.; Thiem, V.D.; Canh, D.G.; Chaicumpa, W.; Agtini, M.D.; Hossain, A.; et al. A Multicentre Study of Shigella Diarrhoea in Six Asian Countries: Disease Burden, Clinical Manifestations, and Microbiology. PLoS Med. 2006, 3, e353. [Google Scholar] [CrossRef]
- Talukder, K.A.; Islam, Z.; Islam, M.A.; Dutta, D.K.; Safa, A.; Ansaruzzaman, M.; Faruque, A.S.G.; Shahed, S.N.; Nair, G.B.; Sack, D.A. Phenotypic and Genotypic Characterization of Provisional Serotype Shigella flexneri 1c and Clonal Relationships with 1a and 1b Strains Isolated in Bangladesh. J. Clin. Microbiol. 2003, 41, 110–117. [Google Scholar] [CrossRef][Green Version]
- Ferreccio, C.; Prado, V.; Ojeda, A.; Cayyazo, M.; Abrego, P.; Guers, L.; Levine, M.M. Epidemiologic Patterns of Acute Diarrhea and Endemic Shigella Infections in Children in a Poor Periurban Setting in Santiago, Chile. Am. J. Epidemiol. 1991, 134, 614–627. [Google Scholar] [CrossRef]
- Gazi, A.; Mahmud, S.; Fahim, S.M.; Kibria, M.G.; Palit, P.; Islam, R.; Rashid, H.; Das, S.; Mahfuz, M.; Ahmeed, T. Functional Prediction of Hypothetical Proteins from Shigella flexneri and Validation of the Predicted Models by Using ROC Curve Analysis. Genom. Inform. 2018, 16, e26. [Google Scholar] [CrossRef]
- Al-Hasani, K.; Henderson, I.; Sakellaris, H.; Rajakumar, K.; Grant, T.; Nataro, J.P.; Robins-Browne, R.; Adler, B. The sigA Gene Which Is Borne on the she Pathogenicity Island of Shigella flexneri 2a Encodes an Exported Cytopathic Protease Involved in Intestinal Fluid Accumulation. Infect. Immun. 2000, 68, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.; Czeczulin, J.; Eslava, C.; Noriega, F.; Nataro, J.P. Characterization of Pic, a Secreted Protease of Shigella flexneri and Enteroaggregative Escherichia coli. Infect. Immun. 1999, 67, 5587–5596. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Gleason, Y.; Wyckoff, E.E.; Payne, S.M. Identification of Two Shigella flexneri Chromosomal Loci Involved in Intercellular Spreading. Infect. Immun. 1998, 66, 4700–4710. [Google Scholar] [CrossRef] [PubMed]
- Mogull, S.; Runyen-Janecky, L.J.; Hong, M.; Payne, S.M. dksA Is Required for Intercellular Spread of Shigella flexneri via an RpoS-Independent Mechanism. Infect. Immun. 2001, 69, 5742–5751. [Google Scholar] [CrossRef] [PubMed]
- Purdy, G.E.; Payne, S.M. The SHI-3 Iron Transport Island of Shigella boydii 0-1392 Carries the Genes for Aerobactin Synthesis and Transport. J. Bacteriol. 2001, 183, 4176–4182. [Google Scholar] [CrossRef]
- Vokes, S.A.; Reeves, S.A.; Torres, A.; Payne, S.M. The aerobactin iron transport system genes in Shigella flexneri are present within a pathogenicity island. Mol. Microbiol. 1999, 33, 63–73. [Google Scholar] [CrossRef]
- Wei, J.; Goldberg, M.B.; Burland, V.; Venkatesan, M.M.; Deng, W.; Fournier, G.; Mayhew, G.F.; Plunkett, G.; Rose, D.J.; Darling, A.E.; et al. Complete Genome Sequence and Comparative Genomics of Shigella flexneri Serotype 2a Strain 2457T. Infect. Immun. 2003, 71, 2775–2786. [Google Scholar]
- Galperin, M.Y.; Koonin, E.V. ‘Conserved hypothetical’ proteins: Prioritization of targets for experimental study. Nucleic Acids Res. 2004, 32, 5452–5463. [Google Scholar] [CrossRef]
- Hawkins, T.; Kihara, D. Function prediction of uncharacterized proteins. J. Bioinform. Comput. Boil. 2007, 5, 1–30. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.-P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef]
- Da Fonsêca, M.M.; Zaha, A.; Caffarena, E.R.; Vasconcelos, A.T. Structure-based functional inference of hypothetical proteins from Mycoplasma hyopneumoniae. J. Mol. Model. 2011, 18, 1917–1925. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Loewenstein, Y.; Raimondo, M.; Redfern, O.; Watson, J.; Frishman, D.; Linial, M.; Orengo, C.; Thornton, J.M.; Tramontano, A. Protein function annotation by homology-based inference. Genome Boil. 2009, 10, 207. [Google Scholar] [CrossRef] [PubMed]
- Nimrod, G.; Schushan, M.; Steinberg, D.M.; Ben-Tal, N. Detection of Functionally Important Regions in “Hypothetical Proteins” of Known Structure. Structure. 2008, 16, 1755–1763. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.; Shahik, M.; Sohel, N.I.A.; Patwary, M.A. Hasan In silico structural and functional annotation of hypothetical proteins of Vibrio cholerae O139. Genom. Inform. 2015, 13, 53–59. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Rahman, S.; Rubi, S.; Zeya, F.; Kumar, K.; Choudhary, H.; Jamal, M.S.; Kim, J.; Hassan, I. Genome analysis of Chlamydia trachomatis for functional characterization of hypothetical proteins to discover novel drug targets. Int. J. Boil. Macromol. 2017, 96, 234–240. [Google Scholar] [CrossRef]
- Singh, A.; Singal, B.; Nath, O.; Singh, I.K. Functional Annotation and Classification of the Hypothetical Proteins of Neisseria meningitidis H44/76. Am. J. of Biosci. and Bioeng. 2015, 3, 57–64. [Google Scholar]
- Mazandu, G.K.; Mulder, N.J. Function prediction and analysis of mycobacterium tuberculosis hypothetical proteins. Int. J. Mol. Sci. 2012, 13, 7283–7302. [Google Scholar] [CrossRef]
- Shahbaaz, M.; Hassan, I.; Ahmad, F. Functional Annotation of Conserved Hypothetical Proteins from Haemophilus influenzae Rd KW20. PLoS ONE 2013, 8, e84263. [Google Scholar] [CrossRef]
- Sarkar, M.; Maganti, L.; Ghoshal, N.; Dutta, C. In silico quest for putative drug targets in Helicobacter pylori HPAG1: Molecular modeling of candidate enzymes from lipopolysaccharide biosynthesis pathway. J. Mol. Model. 2011, 18, 1855–1866. [Google Scholar] [CrossRef]
- Eisenstein, E.; Gilliland, G.L.; Herzberg, O.; Moult, J.; Orban, J.; Poljak, R.J.; Banerjei, L.; Richardson, D.; Howard, A.J. Biological function made crystal clear—Annotation of hypothetical proteins via structural genomics. Curr. Opin. Biotechnol. 2000, 11, 25–30. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2016, 45, D200–D203. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Anderson, J.B.; Derbyshire, M.K.; DeWeese-Scott, C.; Gonzales, N.R.; Gwadz, M.; Hao, L.; He, S.; Hurwitz, D.I.; Jackson, J.D.; et al. CDD: A conserved domain database for interactive domain family analysis. Nucleic Acids Res. 2006, 35, D237–D240. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.; Punta, M.; Qureshi, M.; Sangrador, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2015, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.L.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-S.; Cheng, C.-W.; Su, W.-C.; Chang, S.-C.; Huang, S.-W.; Hwang, J.-K.; Lu, C.-H. CELLO2GO: A Web Server for Protein subCELlular LOcalization Prediction with Functional Gene Ontology Annotation. PLoS ONE 2014, 9, e99368. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Springer: Berlin, Germany, 2005; pp. 571–607. [Google Scholar]
- Yu, C.-S.; Lin, C.-J.; Hwang, J.-K. Predicting subcellular localization of proteins for Gram-negative bacteria by support vector machines based on n-peptide compositions. Protein Sci. 2004, 13, 1402–1406. [Google Scholar] [CrossRef]
- Yu, N.; Wagner, J.R.; Laird, M.; Melli, G.; Rey, S.; Lo, R.; Dao, P.; Sahinalp, S.C.; Ester, M.; Foster, L.J.; et al. PSORTb 3.0: Improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010, 26, 1608–1615. [Google Scholar] [CrossRef]
- Bhasin, M.; Garg, A.; Raghava, G.P.S. PSLpred: Prediction of subcellular localization of bacterial proteins. Bioinformatics 2005, 21, 2522–2524. [Google Scholar] [CrossRef]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Boil. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Tusnády, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001, 17, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Armenteros, J.J.A.; Tsirigos, K.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Bendtsen, J.D.; Kiemer, L.; Fausbøll, A.; Brunak, S. Non-classical protein secretion in bacteria. BMC Microbiol. 2005, 5, 58. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. VICMpred: An SVM-based method for the prediction of functional proteins of Gram-negative bacteria using amino acid patterns and composition. Genom. Proteom. Bioinform. 2006, 4, 42–47. [Google Scholar] [CrossRef]
- Garg, A.; Gupta, D. VirulentPred: A SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinform. 2008, 9, 62. [Google Scholar] [CrossRef]
- Shanmugham, B.; Pan, A. Identification and Characterization of Potential Therapeutic Candidates in Emerging Human Pathogen Mycobacterium abscessus: A Novel Hierarchical In Silico Approach. PLoS ONE 2013, 8, e59126. [Google Scholar] [CrossRef]
- Maloney, P.C. Bacterial transporters. Curr. Opin. Cell Boil. 1994, 6, 571–582. [Google Scholar] [CrossRef]
- Klein, J.S.; Lewinson, O. Bacterial ATP-driven transporters of transition metals: Physiological roles, mechanisms of action, and roles in bacterial virulence. Metallomics 2011, 3, 1098. [Google Scholar] [CrossRef]
- Jack, D.L.; Yang, N.M.; Saier, M.H. The drug/metabolite transporter superfamily. JBIC J. Boil. Inorg. Chem. 2001, 268, 3620–3639. [Google Scholar] [CrossRef]
- Federle, M.J. Autoinducer-2-based chemical communication in bacteria: Complexities of interspecies signalling. In Bacterial Sensing and Signalling; Karger Publishers: Basel, Switzerland, 2009; Volume 16, pp. 18–32. [Google Scholar]
- Parkinson, J.S. Signal transduction schemes of bacteria. Cell 1993, 73, 857–871. [Google Scholar] [CrossRef]
- Gotoh, Y.; Eguchi, Y.; Watanabe, T.; Okamoto, S.; Doi, A.; Utsumi, R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Curr. Opin. Microbiol. 2010, 13, 232–239. [Google Scholar] [CrossRef]
- Ren, B. Genome-Wide Location and Function of DNA Binding Proteins. Science 2000, 290, 2306–2309. [Google Scholar] [CrossRef]
- Phan, N.Q.; Uebanso, T.; Shimohata, T.; Nakahashi, M.; Mawatari, K.; Takahashi, A. DNA-Binding Protein HU Coordinates Pathogenicity in Vibrio parahaemolyticus. J. Bacteriol. 2015, 197, 2958–2964. [Google Scholar] [CrossRef]
- Ariyachet, C.; Solis, N.V.; Liu, Y.; Prasadarao, N.V.; Filler, S.G.; McBride, A.E. SR-Like RNA-Binding Protein Slr1 Affects Candida albicans Filamentation and Virulence. Infect. Immun. 2013, 81, 1267–1276. [Google Scholar] [CrossRef]
- Kondo, Y.; Ohara, N.; Sato, K.; Yoshimura, M.; Yukitake, H.; Naito, M.; Fujiwara, T.; Nakayama, K. Tetratricopeptide Repeat Protein-Associated Proteins Contribute to the Virulence of Porphyromonas gingivalis. Infect. Immun. 2010, 78, 2846–2856. [Google Scholar]
- Shi, X.-Z.; Feng, X.-W.; Sun, J.-J.; Yang, M.-C.; Jiang-Feng, L.; Zhao, X.-F.; Wang, J.-X. Involvement of a LysM and putative peptidoglycan-binding domain-containing protein in the antibacterial immune response of kuruma shrimp Marsupenaeus japonicus. Fish Shellfish. Immunol. 2016, 54, 489–498. [Google Scholar] [CrossRef]
- Kajimura, J.; Fujiwara, T.; Yamada, S.; Suzawa, Y.; Nishida, T.; Oyamada, Y.; Hayashi, I.; Yamagishi, J.-I.; Komatsuzawa, H.; Sugai, M. Identification and molecular characterization of anN-acetylmuramyl-l-alanine amidase Sle1 involved in cell separation ofStaphylococcus aureus. Mol. Microbiol. 2005, 58, 1087–1101. [Google Scholar] [CrossRef]
- Hayashi, S.; Wu, H.C. Lipoproteins in bacteria. J. Bioenerg. Biomembr. 1990, 22, 451–471. [Google Scholar] [CrossRef]
- Torti, S.V.; Park, J.T. Lipoprotein of Gram-negative bacteria is essential for growth and division. Nature 1976, 263, 323–326. [Google Scholar] [CrossRef]
- Kovacs-Simon, A.; Titball, R.W.; Michell, S.L. Lipoproteins of Bacterial Pathogens. Infect. Immun. 2010, 79, 548–561. [Google Scholar]
- Wilson, M.M.; Bernstein, H.D. Surface-Exposed Lipoproteins: An Emerging Secretion Phenomenon in Gram-Negative Bacteria. Trends Microbiol. 2016, 24, 198–208. [Google Scholar] [CrossRef]
- Salton, M.R. Bacterial membrane proteins. Microbiol. Sci. 1987, 4, 100–105. [Google Scholar]
- Rollauer, S.E.; Sooreshjani, M.A.; Noinaj, N.; Buchanan, S.K. Outer membrane protein biogenesis in Gram-negative bacteria. Philos. Trans. R. Soc. B: Boil. Sci. 2015, 370, 20150023. [Google Scholar] [CrossRef]
- Grandi, G. Bacterial surface proteins and vaccines. F1000 Biol. Rep. 2010, 2, 36. [Google Scholar] [CrossRef]
- Bjornson, H.S. Enzymes Associated with the Survival and Virulence of Gram-Negative Anaerobes. Clin. Infect. Dis. 1984, 6, 21–24. [Google Scholar] [CrossRef]
- Höltje, J.V. From growth to autolysis: The murein hydrolases in Escherichia coli. Arch. Microbiol. 1995, 164, 243–254. [Google Scholar] [CrossRef]
- Okugawa, S.; Moayeri, M.; Pomerantsev, A.P.; Sastalla, I.; Crown, D.; Gupta, P.K.; Leppla, S. Lipoprotein biosynthesis by prolipoprotein diacylglyceryl transferase is required for efficient spore germination and full virulence of Bacillus anthracis. Mol. Microbiol. 2011, 83, 96–109. [Google Scholar] [CrossRef]
- Burk, D.L.; Ghuman, N.; Wybenga-Groot, L.E.; Berghuis, A.M. X-ray structure of the AAC(6′)-Ii antibiotic resistance enzyme at 1.8 A resolution: Examination of oligomeric arrangements in GNAT superfamily members. Protein Sci. 2003, 12, 426–437. [Google Scholar] [CrossRef]
- McQuiston, J.R.; Vemulapalli, R.; Inzana, T.J.; Schurig, G.G.; Sriranganathan, N.; Fritzinger, D.; Hadfield, T.L.; Warren, R.A.; Snellings, N.; Hoover, D.; et al. Genetic Characterization of a Tn5-Disrupted Glycosyltransferase Gene Homolog in Brucella abortus and Its Effect on Lipopolysaccharide Composition and Virulence. Infect. Immun. 1999, 67, 3830–3835. [Google Scholar] [CrossRef]
- Davey, L.; Ng, C.K.W.; Halperin, S.A.; Lee, S.F. Functional Analysis of Paralogous Thiol-disulfide Oxidoreductases in Streptococcus gordonii. J. Boil. Chem. 2013, 288, 16416–16429. [Google Scholar] [CrossRef]
- Springer, M.; Graffe, M.; Butler, J.S.; Grunberg-Manago, M. Genetic definition of the translational operator of the threonine-tRNA ligase gene in Escherichia coli. Proc. Natl. Acad. Sci. USA 1986, 83, 4384–4388. [Google Scholar] [CrossRef]
- Reffuveille, F.; Connil, N.; Sanguinetti, M.; Posteraro, B.; Chevalier, S.; Auffray, Y.; Rincé, A. Involvement of Peptidylprolyl cis/trans Isomerases in Enterococcus faecalis Virulence. Infect. Immun. 2012, 80, 1728–1735. [Google Scholar] [CrossRef]
- Ronson, C.; Nixon, B.; Ausubel, F.M. Conserved domains in bacterial regulatory proteins that respond to environmental stimuli. Cell 1987, 49, 579–581. [Google Scholar] [CrossRef]
- Hahn, J.; Inamine, G.; Kozlov, Y.; Dubnau, D. Characterization of comE, a late competence operon of Bacillus subtilis required for the binding and uptake of transforming DNA. Mol. Microbiol. 1993, 10, 99–111. [Google Scholar] [CrossRef]
- Green, J.B.; Lower, R.P.J.; Young, J.P.W. The NfeD Protein Family and Its Conserved Gene Neighbours Throughout Prokaryotes: Functional Implications for Stomatin-Like Proteins. J. Mol. Evol. 2009, 69, 657–667. [Google Scholar] [CrossRef]
- Molina-Henares, A.J.; Krell, T.; Guazzaroni, M.E.; Segura, A.; Ramos, J. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol. Rev. 2006, 30, 157–186. [Google Scholar] [CrossRef]
- Vanderpool, C.K.; Gottesman, S. The Novel Transcription Factor SgrR Coordinates the Response to Glucose-Phosphate Stress. J. Bacteriol. 2007, 189, 2238–2248. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, T.; Verma, N.K. Functional Annotation and Curation of Hypothetical Proteins Present in A Newly Emerged Serotype 1c of Shigella flexneri: Emphasis on Selecting Targets for Virulence and Vaccine Design Studies. Genes 2020, 11, 340. https://doi.org/10.3390/genes11030340
Sen T, Verma NK. Functional Annotation and Curation of Hypothetical Proteins Present in A Newly Emerged Serotype 1c of Shigella flexneri: Emphasis on Selecting Targets for Virulence and Vaccine Design Studies. Genes. 2020; 11(3):340. https://doi.org/10.3390/genes11030340
Chicago/Turabian StyleSen, Tanuka, and Naresh K. Verma. 2020. "Functional Annotation and Curation of Hypothetical Proteins Present in A Newly Emerged Serotype 1c of Shigella flexneri: Emphasis on Selecting Targets for Virulence and Vaccine Design Studies" Genes 11, no. 3: 340. https://doi.org/10.3390/genes11030340
APA StyleSen, T., & Verma, N. K. (2020). Functional Annotation and Curation of Hypothetical Proteins Present in A Newly Emerged Serotype 1c of Shigella flexneri: Emphasis on Selecting Targets for Virulence and Vaccine Design Studies. Genes, 11(3), 340. https://doi.org/10.3390/genes11030340