The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor


Abstract
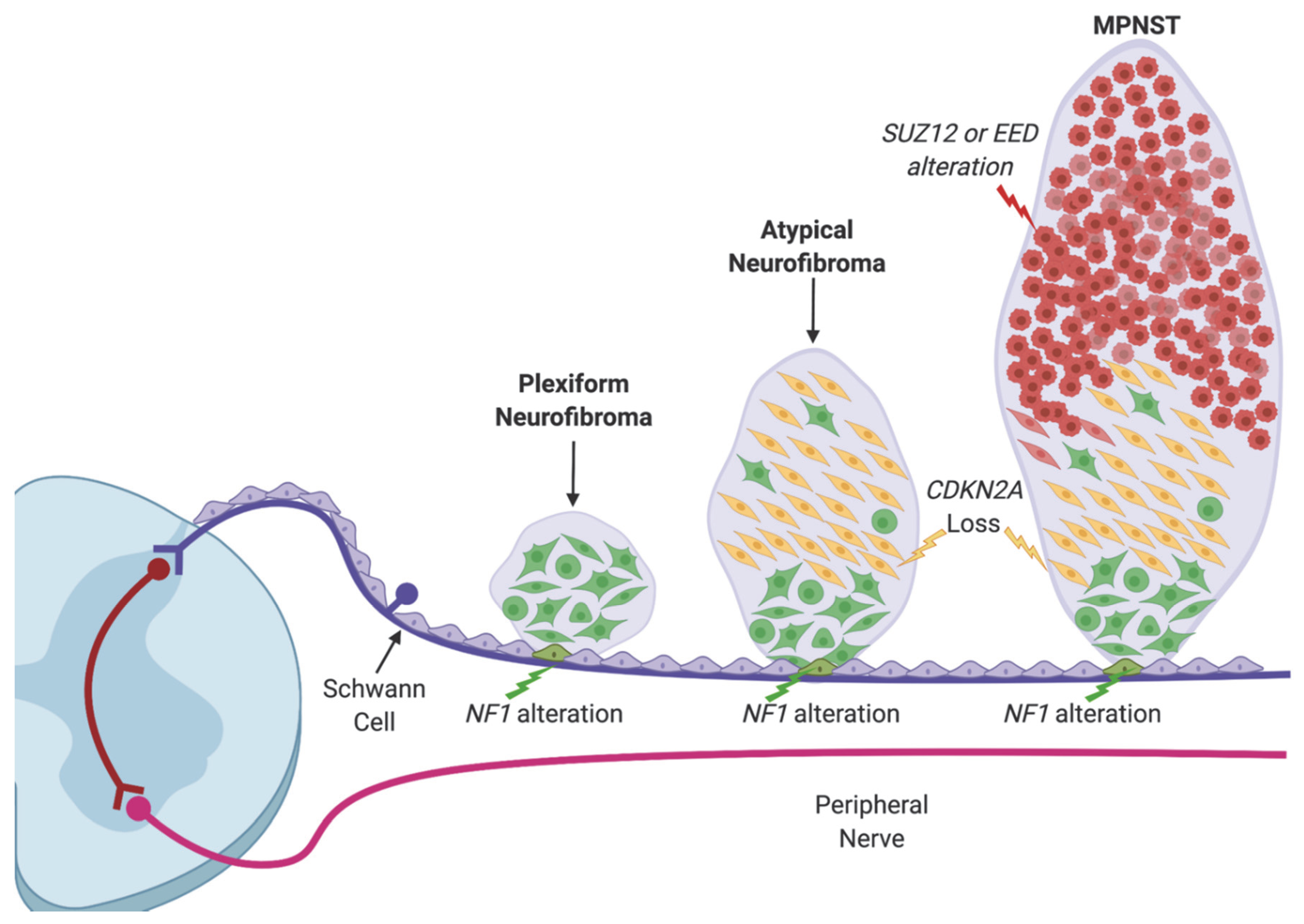
1. Introduction
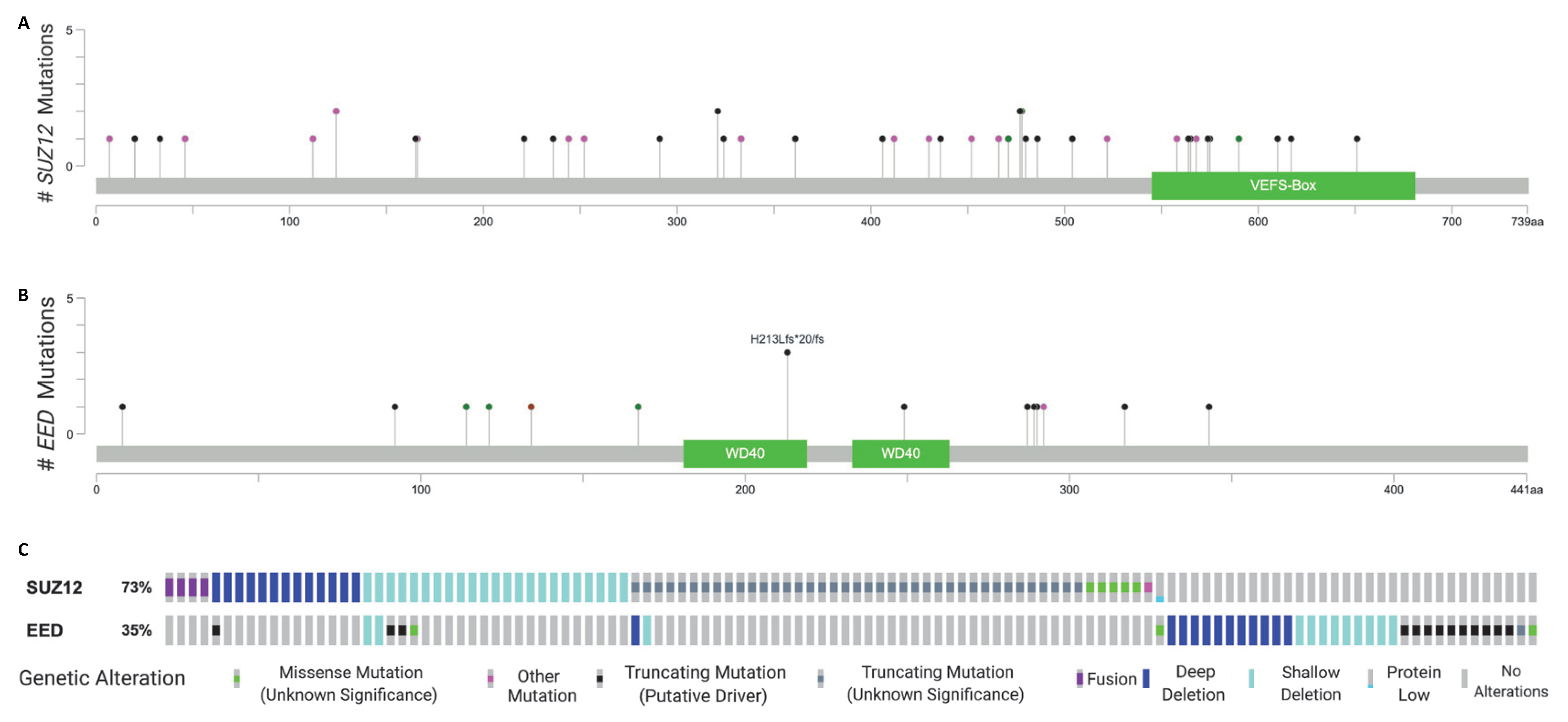
2. Recurrent Mutations in EED and SUZ12 in MPNST
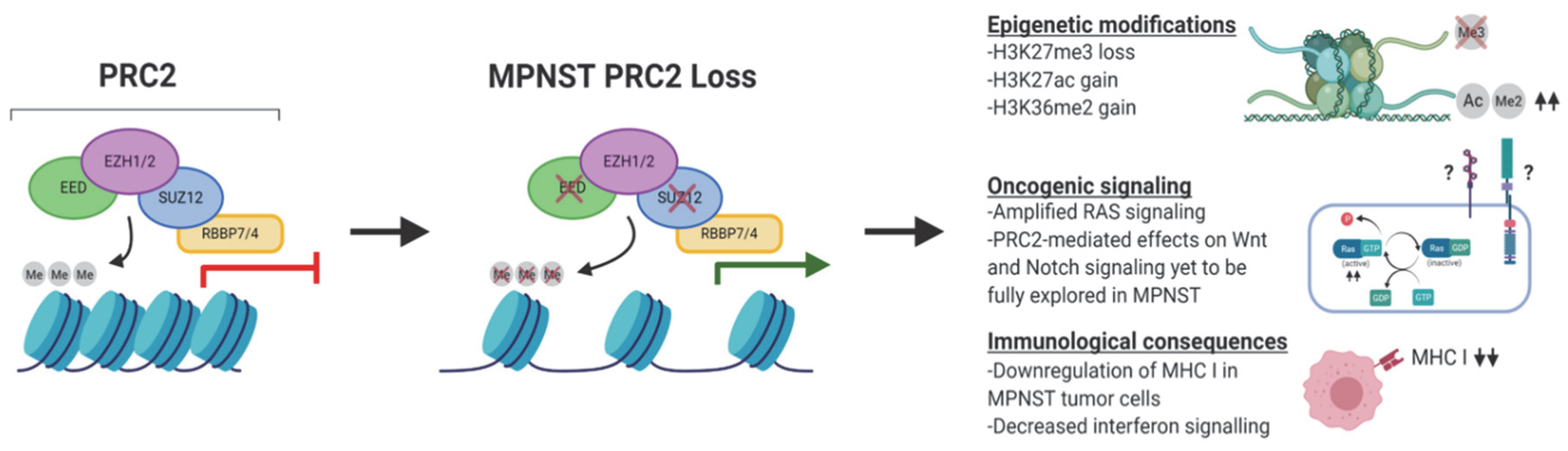
3. The Biochemical, Epigenetic, and Transcriptomic Consequences of PRC2 Loss in MPNST
4. The Role of PRC2 in Schwann Cell Development and Nerve Injury
5. Consequences of PRC2 Loss on Oncogenic Signaling in MPNST
5.1. PRC2 Loss and RAS Signaling
5.2. PRC2 Loss and Wnt Signaling
5.3. PRC2 Loss and Notch Signaling
6. Consequences of PRC2 Loss on Tumor Immune Surveillance in MPNST
7. Establishment of Preclinical Modeling of the PRC2 Loss in MPNST as a Pathway to Clinical Translation
8. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ferner, R.E. Neurofibromatosis 1 and neurofibromatosis 2: A twenty first century perspective. Lancet Neurol. 2007, 6, 340–351. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Publ. Group Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- Martin, G.A.; Viskochil, D.; Bollag, G.; McCabe, P.C.; Crosier, W.J.; Haubruck, H.; Conroy, L.; Clark, R.; O’Connell, P.; Cawthon, R.M. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990, 63, 843–849. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Jett, K.; Friedman, J.M. Clinical and genetic aspects of neurofibromatosis 1. Genet. Med. 2010, 12, 1–11. [Google Scholar] [CrossRef]
- Ferner, R.E.; Huson, S.M.; Thomas, N.; Moss, C.; Willshaw, H.; Evans, D.G.; Upadhyaya, M.; Towers, R.; Gleeson, M.; Steiger, C.; et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet. 2007, 44, 81–88. [Google Scholar] [CrossRef]
- Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch. Neurol. 1988, 45, 575–578. [Google Scholar]
- Evans, D.G.R.; Salvador, H.; Chang, V.Y.; Erez, A.; Voss, S.D.; Druker, H.; Scott, H.S.; Tabori, U. Cancer and Central Nervous System Tumor Surveillance in Pediatric Neurofibromatosis 2 and Related Disorders. Clin. Cancer Res. 2017, 23, e54–e61. [Google Scholar] [CrossRef]
- Bates, J.E.; Peterson, C.R.; Dhakal, S.; Giampoli, E.J.; Constine, L.S. Malignant peripheral nerve sheath tumors (MPNST): A SEER analysis of incidence across the age spectrum and therapeutic interventions in the pediatric population. Pediatric Blood Cancer 2014, 61, 1955–1960. [Google Scholar] [CrossRef]
- Evans, D.G.R.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef]
- Widemann, B.C. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr. Oncol. Rep. 2009, 11, 322–328. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 1–5. [Google Scholar] [CrossRef]
- Cichowski, K.; Jacks, T. NF1 Tumor Suppressor Gene Function. Cell 2001, 104, 593–604. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Dahiya, S.; Miller, C.A.; Li, T.; Fulton, R.S.; Zhang, X.; McDonald, S.; DeSchryver, K.; Duncavage, E.J.; Walrath, J.; et al. Whole Exome Sequencing Reveals the Order of Genetic Changes during Malignant Transformation and Metastasis in a Single Patient with NF1-plexiform Neurofibroma. Clin. Cancer Res. 2015, 21, 4201–4211. [Google Scholar] [CrossRef]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define pre-malignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol. 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Uusitalo, E.; Rantanen, M.; Kallionpää, R.A.; Pöyhönen, M.; Leppävirta, J.; Ylä-Outinen, H.; Riccardi, V.M.; Pukkala, E.; Pitkäniemi, J.; Peltonen, S.; et al. Distinctive Cancer Associations in Patients with Neurofibromatosis Type 1. J. Clin. Oncol. 2016, 34, 1978–1986. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto-Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Rev. Cancer 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2016, 514, 247–251. [Google Scholar] [CrossRef]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Rev. Cancer 2014, 46, 1170–1172. [Google Scholar] [CrossRef]
- Sohier, P.; Luscan, A.; Lloyd, A.; Ashelford, K.; Laurendeau, I.; Briand-Suleau, A.; Vidaud, D.; Ortonne, N.; Pasmant, E.; Upadhyaya, M. Confirmation of mutation landscape of NF1-associated malignant peripheral nerve sheath tumors. Genes Chromosomes Cancer 2017, 56, 421–426. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Electronic address: Elizabeth.demicco@sinaihealthsystem.ca; Cancer Genome Atlas Research Network Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Kim, K.H.; Roberts, C.W.M. Targeting EZH2 in cancer. Nat. Med. 2016, 22, 128–134. [Google Scholar] [CrossRef]
- Wassef, M.; Luscan, A.; Aflaki, S.; Zielinski, D.; Jansen, P.W.T.C.; Baymaz, H.I.; Battistella, A.; Kersouani, C.; Servant, N.; Wallace, M.R.; et al. EZH1/2 function mostly within canonical PRC2 and exhibit proliferation-dependent redundancy that shapes mutational signatures in cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 6075–6080. [Google Scholar] [CrossRef]
- Sneeringer, C.J.; Scott, M.P.; Kuntz, K.W.; Knutson, S.K.; Pollock, R.M.; Richon, V.M.; Copeland, R.A. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 20980–20985. [Google Scholar] [CrossRef]
- Li, X.; Gonzalez, M.E.; Toy, K.; Filzen, T.; Merajver, S.D.; Kleer, C.G. Targeted Overexpression of EZH2 in the Mammary Gland Disrupts Ductal Morphogenesis and Causes Epithelial Hyperplasia. Am. J. Pathol. 2009, 175, 1246–1254. [Google Scholar] [CrossRef]
- Karanikolas, B.D.W.; Figueiredo, M.L.; Wu, L. Polycomb Group Protein Enhancer of Zeste 2 Is an Oncogene That Promotes the Neoplastic Transformation of a Benign Prostatic Epithelial Cell Line. Mol. Cancer Res. 2009, 7, 1456–1465. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 11606–11611. [Google Scholar] [CrossRef]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181. [Google Scholar] [CrossRef]
- Yap, D.B.; Chu, J.; Berg, T.; Schapira, M.; Cheng, S.W.G.; Moradian, A.; Morin, R.D.; Mungall, A.J.; Meissner, B.; Boyle, M.; et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood 2011, 117, 2451–2459. [Google Scholar] [CrossRef]
- Souroullas, G.P.; Jeck, W.R.; Parker, J.S.; Simon, J.M.; Liu, J.-Y.; Paulk, J.; Xiong, J.; Clark, K.S.; Fedoriw, Y.; Qi, J.; et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat. Med. 2019, 22, 632. [Google Scholar] [CrossRef]
- McCabe, M.T.; Graves, A.P.; Ganji, G.; Diaz, E.; Halsey, W.S.; Jiang, Y.; Smitheman, K.N.; Ott, H.M.; Pappalardi, M.B.; Allen, K.E.; et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc. Natl. Acad. Sci. USA 2012, 109, 2989–2994. [Google Scholar] [CrossRef]
- Majer, C.R.; Jin, L.; Scott, M.P.; Knutson, S.K.; Kuntz, K.W.; Keilhack, H.; Smith, J.J.; Moyer, M.P.; Richon, V.M.; Copeland, R.A.; et al. A687V EZH2 is a gain-of-function mutation found in lymphoma patients. FEBS Lett. 2012, 586, 3448–3451. [Google Scholar] [CrossRef]
- Zhan, F.; Hardin, J.; Kordsmeier, B.; Bumm, K.; Zheng, M.; Tian, E.; Sanderson, R.; Yang, Y.; Wilson, C.; Zangari, M.; et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood 2002, 99, 1745–1757. [Google Scholar] [CrossRef]
- Croonquist, P.A.; Van Ness, B. The polycomb group protein enhancer of zeste homolog 2 (EZH2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene 2005, 24, 6269–6280. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef]
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 Expression Is Associated With High Proliferation Rate and Aggressive Tumor Subgroups in Cutaneous Melanoma and Cancers of the Endometrium, Prostate, and Breast. J. Clin. Oncol. 2006, 24, 268–273. [Google Scholar] [CrossRef]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- The St. Jude Children’s Researchs Hospital–Washington University Pediatric Cancer Genome Project. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Rev. Cancer 2014, 46, 444–450. [Google Scholar]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 is a potential therapeutic target for H3K27M-mutant pediatric gliomas. Nat. Med. 2017, 23, 484. [Google Scholar] [CrossRef]
- Margueron, R.; Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef]
- Müller, J.; Verrijzer, P. Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 2009, 19, 150–158. [Google Scholar] [CrossRef]
- Simon, J.A.; Kingston, R.E. Mechanisms of Polycomb gene silencing: Knowns and unknowns. Nat. Rev. Mol. Cell Biol. 2009, 10, 697–708. [Google Scholar] [CrossRef]
- Cao, R.; Zhang, Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004, 14, 155–164. [Google Scholar] [CrossRef]
- Bracken, A.P. Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stützer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef]
- Højfeldt, J.W.; Laugesen, A.; Willumsen, B.M.; Damhofer, H.; Hedehus, L.; Tvardovskiy, A.; Mohammad, F.; Jensen, O.N.; Helin, K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018, 25, 225–232. [Google Scholar] [CrossRef]
- Jung, H.R.; Pasini, D.; Helin, K.; Jensen, O.N. Quantitative Mass Spectrometry of Histones H3.2 and H3.3 in Suz12-deficient Mouse Embryonic Stem Cells Reveals Distinct, Dynamic Post-translational Modifications at Lys-27 and Lys-36. Mol. Cell. Proteom. 2010, 9, 838–850. [Google Scholar] [CrossRef]
- Youmans, D.T.; Schmidt, J.C.; Cech, T.R. Live-cell imaging reveals the dynamics of PRC2 and recruitment to chromatin by SUZ12-associated subunits. Genes Dev. 2018, 32, 794–805. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Marchione, D.M.; Lisby, A.; Viaene, A.N.; Santi, M.; Nasrallah, M.; Wang, L.-P.; Williams, E.A.; Larque, A.B.; Chebib, I.; Garcia, B.A.; et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod. Pathol. 2019, 32, 1434–1446. [Google Scholar] [CrossRef]
- Pekmezci, M.; Cuevas-Ocampo, A.K.; Perry, A.; Horvai, A.E. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod. Pathol. 2017, 30, 1710–1719. [Google Scholar] [CrossRef]
- Asano, N.; Yoshida, A.; Ichikawa, H.; Mori, T.; Nakamura, M.; Kawai, A.; Hiraoka, N. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology 2017, 70, 385–393. [Google Scholar] [CrossRef]
- Le Guellec, S.; Macagno, N.; Velasco, V.; Lamant, L.; Lae, M.; Filleron, T.; Malissen, N.; Cassagnau, E.; Terrier, P.; Chevreau, C.; et al. Loss of H3K27 trimethylation is not suitable for distinguishing malignant peripheral nerve sheath tumor from melanoma: A study of 387 cases including mimicking lesions. Mod. Pathol. 2017, 30, 1677–1687. [Google Scholar] [CrossRef]
- Otsuka, H.; Kohashi, K.; Yoshimoto, M.; Ishihara, S.; Toda, Y.; Yamada, Y.; Yamamoto, H.; Nakashima, Y.; Oda, Y. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol. Res. Pract. 2018, 214, 417–425. [Google Scholar] [CrossRef]
- Cleven, A.H.G.; Sannaa Al, G.A.; Bruijn, I.B.-D.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.-L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 582–590. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; Fletcher, C.D.; Hornick, J.L. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod. Pathol. 2016, 29, 4–13. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489. [Google Scholar] [CrossRef]
- Wojcik, J.B.; Marchione, D.M.; Sidoli, S.; Djedid, A.; Lisby, A.; Majewski, J.; Garcia, B.A. Epigenomic reordering induced by Polycomb loss drives oncogenesis but leads to therapeutic vulnerabilities in malignant peripheral nerve sheath tumors. Cancer Res. 2019, 79, 3205–3219. [Google Scholar] [CrossRef]
- Bell, O.; Wirbelauer, C.; Hild, M.; Scharf, A.N.D.; Schwaiger, M.; MacAlpine, D.M.; Zilbermann, F.; van Leeuwen, F.; Bell, S.P.; Imhof, A.; et al. Localized H3K36 methylation states define histone H4K16 acetylation during transcriptional elongation in Drosophila. EMBO J. 2007, 26, 4974–4984. [Google Scholar] [CrossRef]
- Turberfield, A.H.; Kondo, T.; Nakayama, M.; Koseki, Y.; King, H.W.; Koseki, H.; Klose, R.J. KDM2 proteins constrain transcription from CpG island gene promoters independently of their histone demethylase activity. Nucleic Acids Res. 2019, 47, 9005–9023. [Google Scholar] [CrossRef]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef]
- Yuan, W.; Xu, M.; Huang, C.; Liu, N.; Chen, S.; Zhu, B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J. Biol. Chem. 2011, 286, 7983–7989. [Google Scholar] [CrossRef]
- Xie, W.; Schultz, M.D.; Lister, R.; Hou, Z.; Rajagopal, N.; Ray, P.; Whitaker, J.W.; Tian, S.; Hawkins, R.D.; Leung, D.; et al. Epigenomic Analysis of Multilineage Differentiation of Human Embryonic Stem Cells. Cell 2013, 153, 1134–1148. [Google Scholar] [CrossRef]
- Long, H.K.; Sims, D.; Heger, A.; Blackledge, N.P.; Kutter, C.; Wright, M.L.; Grützner, F.; Odom, D.T.; Patient, R.; Ponting, C.P.; et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. Elife 2013, 2, e00348. [Google Scholar] [CrossRef]
- Jeong, M.; Sun, D.; Luo, M.; Huang, Y.; Challen, G.A.; Rodriguez, B.; Zhang, X.; Chavez, L.; Wang, H.; Hannah, R.; et al. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Rev. Cancer 2013, 46, 17–23. [Google Scholar] [CrossRef]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-Interacting Proteins Regulated by DNA and Histone Methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef]
- Wu, H.; Coskun, V.; Tao, J.; Xie, W.; Ge, W.; Yoshikawa, K.; Li, E.; Zhang, Y.; Sun, Y.E. Dnmt3a-Dependent Nonpromoter DNA Methylation Facilitates Transcription of Neurogenic Genes. Science 2010, 329, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.; Dienstbier, M.; Hassan, R.; Schermelleh, L.; Sharif, J.; Blackledge, N.P.; De Marco, V.; Elderkin, S.; Koseki, H.; Klose, R.; et al. Targeting Polycomb to Pericentric Heterochromatin in Embryonic Stem Cells Reveals a Role for H2AK119u1 in PRC2 Recruitment. Cell Rep. 2014, 7, 1456–1470. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, H.; Wang, Q.; Zhou, C.; Wei, L.; Liu, X.; Zhang, W.; Zhang, Y.; Du, Z.; Wang, X.; et al. Genome-wide analyses reveal a role of Polycomb in promoting hypomethylation of DNA methylation valleys. Genome Biol. 2018, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef]
- Cao, R.; Tsukada, Y.-I.; Zhang, Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell 2005, 20, 845–854. [Google Scholar] [CrossRef]
- Pengelly, A.R.; Copur, Ö.; Jäckle, H.; Herzig, A.; Müller, J. A Histone Mutant Reproduces the Phenotype Caused by Loss of Histone-Modifying Factor Polycomb. Science 2013, 339, 698–699. [Google Scholar] [CrossRef]
- Francis, N.J.; Kingston, R.E. Mechanisms of transcriptional memory. Nat. Rev. Mol. Cell Biol. 2001, 2, 409–421. [Google Scholar] [CrossRef]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 2002, 298, 1039–1043. [Google Scholar] [CrossRef]
- Shao, Z.; Raible, F.; Mollaaghababa, R.; Guyon, J.R.; Wu, C.T.; Bender, W.; Kingston, R.E. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 1999, 98, 37–46. [Google Scholar] [CrossRef]
- Kalb, R.; Latwiel, S.; Baymaz, H.I.; Jansen, P.W.T.C.; Müller, C.W.; Vermeulen, M.; Müller, J. Histone H2A monoubiquitination promotes histone H3 methylation in Polycomb repression. Nat. Struct. Mol. Biol. 2014, 21, 569–571. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef]
- Gupta, G.; Mammis, A.; Maniker, A. Malignant peripheral nerve sheath tumors. Neurosurg. Clin. N. Am. 2008, 19, 533–543. [Google Scholar] [CrossRef]
- Chen, Z.; Liu, C.; Patel, A.J.; Liao, C.-P.; Wang, Y.; Le, L.Q. Cells of Origin in the Embryonic Nerve Roots for NF1-Associated Plexiform Neurofibroma. Cancer Cell 2014, 26, 695–706. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The Success and Failure of the Schwann Cell Response to Nerve Injury. Front. Cell. Neurosci. 2019, 13, 16. [Google Scholar] [CrossRef]
- Clements, M.P.; Byrne, E.; Camarillo Guerrero, L.F.; Cattin, A.-L.; Zakka, L.; Ashraf, A.; Burden, J.J.; Khadayate, S.; Lloyd, A.C.; Marguerat, S.; et al. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 2017, 96, 98–114. [Google Scholar] [CrossRef]
- Wekerle, H.; Schwab, M.; Linington, C.; Meyermann, R. Antigen presentation in the peripheral nervous system: Schwann cells present endogenous myelin autoantigens to lymphocytes. Eur. J. Immunol. 1986, 16, 1551–1557. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. The repair Schwann cell and its function in regenerating nerves. J. Physiol. 2016, 594, 3521–3531. [Google Scholar] [CrossRef]
- Mirsky, R.; Jessen, K.R. Schwann cell development, differentiation and myelination. Curr. Opin. Neurobiol. 1996, 6, 89–96. [Google Scholar] [CrossRef]
- Castelnovo, L.; Bonalume, V.; Melfi, S.; Ballabio, M.; Colleoni, D.; Magnaghi, V. Schwann cell development, maturation and regeneration: A focus on classic and emerging intracellular signaling pathways. Neural Regen. Res. 2017, 12, 1013. [Google Scholar]
- Woodhoo, A.; Sommer, L. Development of the Schwann cell lineage: From the neural crest to the myelinated nerve. Glia 2008, 56, 1481–1490. [Google Scholar] [CrossRef]
- Jessen, K.R.; Mirsky, R. Schwann Cell Precursors; Multipotent Glial Cells in Embryonic Nerves. Front. Mol. Neurosci. 2019, 12, 366. [Google Scholar] [CrossRef]
- Furlan, A.; Adameyko, I. Schwann cell precursor_ a neural crest cell in disguise? Dev. Biol. 2018, 444, S25–S35. [Google Scholar] [CrossRef]
- Stolt, C.C.; Wegner, M. Schwann cells and their transcriptional network: Evolution of key regulators of peripheral myelination. Brain Res. 2016, 1641, 101–110. [Google Scholar] [CrossRef]
- Jacob, C. ScienceDirect Chromatin-remodeling enzymes in control of Schwann cell development, maintenance and plasticity. Curr. Opin. Neurobiol. 2017, 47, 24–30. [Google Scholar] [CrossRef]
- Ma, K.H.; Svaren, J. Epigenetic Control of Schwann Cells. Neuroscientist 2018, 24, 627–638. [Google Scholar] [CrossRef]
- Ness, J.K.; Skiles, A.A.; Yap, E.H.; Fajardo, E.J.; Fiser, A.; Tapinos, N. Nuc-ErbB3 regulates H3K27me3 levels and HMT activity to establish epigenetic repression during peripheral myelination. Glia 2016, 64, 977–992. [Google Scholar] [CrossRef][Green Version]
- Ma, K.H.; Hung, H.A.; Srinivasan, R.; Xie, H.; Orkin, S.H.; Svaren, J. Regulation of Peripheral Nerve Myelin Maintenance by Gene Repression through Polycomb Repressive Complex 2. J. Neurosci. 2015, 35, 8640–8652. [Google Scholar] [CrossRef]
- Heinen, A.; Tzekova, N.; Graffmann, N.; Torres, K.J.; Uhrberg, M.; Hartung, H.P.; Küry, P. Histone methyltransferase enhancer of zeste homolog 2 regulates Schwann cell differentiation. Glia 2012, 60, 1696–1708. [Google Scholar] [CrossRef]
- Chen, Z.-L.; Yu, W.-M.; Strickland, S. Peripheral Regeneration. Annu. Rev. Neurosci. 2007, 30, 209–233. [Google Scholar] [CrossRef]
- Fontana, X.; Hristova, M.; Da Costa, C.; Patodia, S.; Thei, L.; Makwana, M.; Spencer-Dene, B.; Latouche, M.; Mirsky, R.; Jessen, K.R.; et al. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J. Cell Biol. 2012, 198, 127–141. [Google Scholar] [CrossRef]
- Hirota, H.; Kiyama, H.; Kishimoto, T.; Taga, T. Accelerated Nerve Regeneration in Mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J. Exp. Med. 1996, 183, 2627–2634. [Google Scholar] [CrossRef]
- Rotshenker, S. Wallerian degeneration: The innate-immune response to traumatic nerve injury. J. Neuroinflammation 2011, 8, 109. [Google Scholar] [CrossRef]
- Cattin, A.-L.; Burden, J.J.; Van Emmenis, L.; Mackenzie, F.E.; Hoving, J.J.A.; Garcia Calavia, N.; Guo, Y.; McLaughlin, M.; Rosenberg, L.H.; Quereda, V.; et al. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 2015, 162, 1127–1139. [Google Scholar] [CrossRef]
- Stoll, G.; Müller, H.W. Nerve Injury, Axonal Degeneration and Neural Regeneration: Basic Insights. Brain Pathol. 1999, 9, 313–325. [Google Scholar] [CrossRef]
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502. [Google Scholar] [CrossRef]
- Ma, K.H.; Hung, H.A.; Svaren, J. Epigenomic Regulation of Schwann Cell Reprogramming in Peripheral Nerve Injury. J. Neurosci. 2016, 36, 9135–9147. [Google Scholar] [CrossRef]
- Ribeiro, S.; Napoli, I.; White, I.J.; Parrinello, S.; Flanagan, A.M.; Suter, U.; Parada, L.F.; Lloyd, A.C. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell Rep. 2013, 5, 126–136. [Google Scholar] [CrossRef]
- Korfhage, J.; Lombard, D.B. Malignant Peripheral Nerve Sheath Tumors: From Epigenome to Bedside. Mol. Cancer Res. 2019, 17, 1417–1428. [Google Scholar] [CrossRef]
- Bottillo, I.; Ahlquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1–Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Kim, A.; Stewart, D.R.; Reilly, K.M.; Viskochil, D.; Miettinen, M.M.; Widemann, B.C. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017, 2017, 7429697. [Google Scholar] [CrossRef]
- Carroll, S.L. The Challenge of Cancer Genomics in Rare Nervous System Neoplasms: Malignant Peripheral Nerve Sheath Tumors as a Paradigm for Cross-Species Comparative Oncogenomics. Am. J. Pathol. 2016, 186, 464–477. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-Enhancers in the Control of Cell Identity and Disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Adhikari, A.; Davie, J. JARID2 and the PRC2 complex regulate skeletal muscle differentiation through regulation of canonical Wnt signaling. Epigenet. Chromatin 2018, 11, 1–20. [Google Scholar] [CrossRef]
- Mirzamohammadi, F.; Papaioannou, G.; Inloes, J.B.; Rankin, E.B.; Xie, H.; Schipani, E.; Orkin, S.H.; Kobayashi, T. Polycomb repressive complex 2 regulates skeletal growth by suppressing Wnt and TGF-β signalling. Nat. Commun. 2016, 7, 12047. [Google Scholar] [CrossRef]
- Wang, L.; Jin, Q.; Lee, J.-E.; Su, I.-H.; Ge, K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 7317–7322. [Google Scholar] [CrossRef]
- Rothberg, J.L.M.; Maganti, H.B.; Jrade, H.; Porter, C.J.; Palidwor, G.A.; Cafariello, C.; Battaion, H.L.; Khan, S.T.; Perkins, T.J.; Paulson, R.F.; et al. Mtf2-PRC2 control of canonical Wnt signaling is required for definitive erythropoiesis. Cell Discov. 2018, 4, 1–16. [Google Scholar] [CrossRef]
- Oittinen, M.; Popp, A.; Kurppa, K.; Lindfors, K.; Mäki, M.; Kaikkonen, M.U.; Viiri, K. Polycomb Repressive Complex 2 Enacts Wnt Signaling in Intestinal Homeostasis and Contributes to the Instigation of Stemness in Diseases Entailing Epithelial Hyperplasia or Neoplasia. Stem Cells 2017, 35, 445–457. [Google Scholar] [CrossRef]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef]
- Chen, C.; Zhao, M.; Tian, A.; Zhang, X.; Yao, Z.; Ma, X. Aberrant activation of Wnt/β-catenin signaling drives proliferation of bone sarcoma cells. Oncotarget 2015, 6, 17570–17583. [Google Scholar] [CrossRef]
- Üren, A.; Wolf, V.; Sun, Y.F.; Azari, A.; Rubin, J.S.; Toretsky, J.A. Wnt/Frizzled signaling in Ewing sarcoma. Pediatric Blood Cancer 2004, 43, 243–249. [Google Scholar] [CrossRef]
- Abhinav Adhikari, J.D. Wnt deregulation in rhabdomyosarcoma. Stem Cell Investig. 2019, 6, 13. [Google Scholar] [CrossRef]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/β-catenin Signaling Drives Human Schwann Cell Transformation, Progression, and Tumor Maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef]
- Luscan, A.; Shackleford, G.; Masliah-Planchon, J.; Laurendeau, I.; Ortonne, N.; Varin, J.; Lallemand, F.; Leroy, K.; Dumaine, V.; Hivelin, M.; et al. The Activation of the WNT Signaling Pathway Is a Hallmark in Neurofibromatosis Type 1 Tumorigenesis. Clin. Cancer Res. 2014, 20, 358–371. [Google Scholar] [CrossRef]
- Hu, P.; Chu, J.; Wu, Y.; Sun, L.; Lv, X.; Zhu, Y.; Li, J.; Guo, Q.; Gong, C.; Liu, B.; et al. NBAT1 suppresses breast cancer metastasis by regulating DKK1 via PRC2. Oncotarget 2015, 6, 32410–32425. [Google Scholar] [CrossRef]
- Nakagawa, M.; Fujita, S.; Katsumoto, T.; Yamagata, K.; Ogawara, Y.; Hattori, A.; Kagiyama, Y.; Honma, D.; Araki, K.; Inoue, T.; et al. Dual inhibition of enhancer of zeste homolog 1/2 overactivates WNT signaling to deplete cancer stem cells in multiple myeloma. Cancer Sci. 2019, 110, 194–208. [Google Scholar] [CrossRef]
- Serresi, M.; Gargiulo, G.; Proost, N.; Siteur, B.; Cesaroni, M.; Koppens, M.; Xie, H.; Sutherland, K.D.; Hulsman, D.; Citterio, E.; et al. Polycomb Repressive Complex 2 Is a Barrier to KRAS- Driven Inflammation and Epithelial-Mesenchymal Transition in Non-Small-Cell Lung Cancer. Cancer Cell 2016, 29, 17–31. [Google Scholar] [CrossRef]
- Tosello, V.; Ferrando, A.A. The NOTCH signaling pathway: Role in the pathogenesis of T-cell acute lymphoblastic leukemia and implication for therapy. Ther. Adv. Hematol. 2013, 4, 199–210. [Google Scholar] [CrossRef]
- Gordon, W.R.; Arnett, K.L.; Blacklow, S.C. The molecular logic of Notch signaling–A structural and biochemical perspective. J. Cell Sci. 2008, 121, 3109–3119. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Kopan, R.; Ilagan, M.X.G. The Canonical Notch Signaling Pathway: Unfolding the Activation Mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef]
- Kovall, R.A. More complicated than it looks: Assembly of Notch pathway transcription complexes. Oncogene 2008, 27, 5099–5109. [Google Scholar] [CrossRef]
- Han, X.; Ranganathan, P.; Tzimas, C.; Weaver, K.L.; Jin, K.; Astudillo, L.; Zhou, W.; Zhu, X.; Li, B.; Robbins, D.J.; et al. Notch Represses Transcription by PRC2 Recruitment to the Ternary Complex. Mol. Cancer Res. 2017, 15, 1173–1183. [Google Scholar] [CrossRef]
- Weijzen, S.; Rizzo, P.; Braid, M.; Vaishnav, R.; Jonkheer, S.M.; Zlobin, A.; Osborne, B.A.; Gottipati, S.; Aster, J.C.; Hahn, W.C.; et al. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat. Med. 2002, 8, 979–986. [Google Scholar] [CrossRef]
- Li, Y.; Rao, P.K.; Wen, R.; Song, Y.; Muir, D.; Wallace, P.; van Horne, S.J.; Tennekoon, G.I.; Kadesch, T. Notch and Schwann cell transformation. Oncogene 2004, 23, 1146–1152. [Google Scholar] [CrossRef]
- Burr, M.L.; Sparbier, C.E.; Chan, K.L.; Chan, Y.-C.; Kersbergen, A.; Lam, E.Y.N.; Azidis-Yates, E.; Vassiliadis, D.; Bell, C.C.; Gilan, O.; et al. An Evolutionarily Conserved Function of Polycomb Silences the MHC Class I Antigen Presentation Pathway and Enables Immune Evasion in Cancer. Cancer Cell 2019, 36, 385–401. [Google Scholar] [CrossRef]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285. [Google Scholar] [CrossRef]
- Zingg, D.; Arenas-Ramirez, N.; Sahin, D.; Rosalia, R.A.; Antunes, A.T.; Haeusel, J.; Sommer, L.; Boyman, O. The Histone Methyltransferase Ezh2 Controls Mechanisms of Adaptive Resistance to Tumor Immunotherapy. Cell Rep. 2017, 20, 854–867. [Google Scholar] [CrossRef]
- Christian, S.L.; Collier, T.W.; Zu, D.; Licursi, M.; Hough, C.M.; Hirasawa, K. Activated Ras/MEK Inhibits the Antiviral Response of Alpha Interferon by Reducing STAT2 Levels. J. Virol. 2009, 83, 6717–6726. [Google Scholar] [CrossRef]
- AbuSara, N.; Razavi, S.; Derwish, L.; Komatsu, Y.; Licursi, M.; Hirasawa, K. Restoration of IRF1-dependent anticancer effects by MEK inhibition in human cancer cells. Cancer Lett. 2015, 357, 575–581. [Google Scholar] [CrossRef]
- Murray, E.K.; Hien, A.; de Vries, G.J.; Forger, N.G. Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 2009, 150, 4241–4247. [Google Scholar] [CrossRef]
- Speert, D.B.; Konkle, A.T.M.; Zup, S.L.; Schwarz, J.M.; Shiroor, C.; Taylor, M.E.; McCarthy, M.M. Focal adhesion kinase and paxillin: Novel regulators of brain sexual differentiation? Endocrinology 2007, 148, 3391–3401. [Google Scholar] [CrossRef][Green Version]
- Amirnasr, A.; Verdijk, R.M.; van Kuijk, P.F.; Taal, W.; Sleijfer, S.; Wiemer, E.A.C. Expression and inhibition of BRD4, EZH2 and TOP2A in neurofibromas and malignant peripheral nerve sheath tumors. PLoS ONE 2017, 12, e0183155. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Kozakewich, H.P.; Hoffer, F.A.; Lage, J.M.; Weidner, N.; Tepper, R.; Pinkus, G.S.; Morton, C.C.; Corson, J.M. Diagnostic Relevance of Clonal Cytogenetic Aberrations in Malignant Soft-Tissue Tumors. N. Engl. J. Med. 2010, 324, 436–443. [Google Scholar] [CrossRef]
- Reynolds, J.E.; Fletcher, J.A.; Lytle, C.H.; Nie, L.; Morton, C.C.; Diehl, S.R. Molecular characterization of a 17q11.2 translocation in a malignant schwannoma cell line. Hum. Genet. 1992, 90, 450–456. [Google Scholar] [CrossRef]
- DeClue, J.E.; Papageorge, A.G.; Fletcher, J.A.; Diehl, S.R.; Ratner, N.; Vass, W.C.; Lowy, D.R. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 1992, 69, 265–273. [Google Scholar] [CrossRef]
- Glover, T.W.; Stein, C.K.; Legius, E.; Andersen, L.B.; Brereton, A.; Johnson, S. Molecular and cytogenetic analysis of tumors in von recklinghausen neurofibromatosis. Genes Chromosom. Cancer 1991, 3, 62–70. [Google Scholar] [CrossRef]
- Yang, K.; Guo, W.; Ren, T.; Huang, Y.; Han, Y.; Zhang, H.; Zhang, J. Knockdown of HMGA2 regulates the level of autophagy via interactions between MSI2 and Beclin1 to inhibit NF1-associated malignant peripheral nerve sheath tumour growth. J. Exp. Clin. Cancer Res. 2019, 38, 1–18. [Google Scholar] [CrossRef]
- Li, H.; Zhang, X.; Fishbein, L.; Kweh, F.; Campbell-Thompson, M.; Perrin, G.Q.; Muir, D.; Wallace, M. Analysis of steroid hormone effects on xenografted human NF1 tumor schwann cells. Cancer Biol. Ther. 2010, 10, 758–764. [Google Scholar] [CrossRef]
- Kahen, E.J.; Brohl, A.; Yu, D.; Welch, D.; Cubitt, C.L.; Lee, J.K.; Chen, Y.; Yoder, S.J.; Teer, J.K.; Zhang, Y.O.; et al. Neurofibromin level directs RAS pathway signaling and mediates sensitivity to targeted agents in malignant peripheral nerve sheath tumors. Oncotarget 2018, 9, 22571–22585. [Google Scholar] [CrossRef]
- Perrin, G.Q.; Fishbein, L.; Thomson, S.A.; Thomas, S.L.; Stephens, K.; Garbern, J.Y.; DeVries, G.H.; Yachnis, A.T.; Wallace, M.R.; Muir, D. Plexiform-like neurofibromas develop in the mouse by intraneural xenograft of an NF1 tumor-derived Schwann cell line. J. Neurosci. Res. 2007, 85, 1347–1357. [Google Scholar] [CrossRef]
- Perrin, G.Q.; Li, H.; Fishbein, L.; Thomson, S.A.; Hwang, M.S.; Scarborough, M.T.; Yachnis, A.T.; Wallace, M.R.; Mareci, T.H.; Muir, D. An orthotopic xenograft model of intraneural NF1 MPNST suggests a potential association between steroid hormones and tumor cell proliferation. Lab. Investig. 2007, 87, 1092–1102. [Google Scholar] [CrossRef]
- Frahm, S.; Mautner, V.-F.; Brems, H.; Legius, E.; Debiec-Rychter, M.; Friedrich, R.E.; Knöfel, W.T.; Peiper, M.; Kluwe, L. Genetic and phenotypic characterization of tumor cells derived from malignant peripheral nerve sheath tumors of neurofibromatosis type 1 patients. Neurobiol. Dis. 2004, 16, 85–91. [Google Scholar] [CrossRef]
- Mahller, Y.Y.; Vaikunth, S.S.; Ripberger, M.C.; Baird, W.H.; Saeki, Y.; Cancelas, J.A.; Crombleholme, T.M.; Cripe, T.P. Tissue Inhibitor of Metalloproteinase-3 via Oncolytic Herpesvirus Inhibits Tumor Growth and Vascular Progenitors. Cancer Res. 2008, 68, 1170–1179. [Google Scholar] [CrossRef]
- Mashour, G.A.; Drissel, S.N.; Frahm, S.; Farassati, F.; Martuza, R.L.; Mautner, V.-F.; Kindler-Röhrborn, A.; Kurtz, A. Differential modulation of malignant peripheral nerve sheath tumor growth by omega-3 and omega-6 fatty acids. Oncogene 2005, 24, 2367–2374. [Google Scholar] [CrossRef]
- Hakozaki, M.; Hojo, H.; Sato, M.; Tajino, T.; Yamada, H.; Kikuchi, S.; Abe, M. Establishment and characterization of a novel human malignant peripheral nerve sheath tumor cell line, FMS-1, that overexpresses epidermal growth factor receptor and cyclooxygenase-2. Virchows Arch. 2009, 455, 517–526. [Google Scholar] [CrossRef]
- Aoki, M.; Nabeshima, K.; Nishio, J.; Ishiguro, M.; Fujita, C.; Koga, K.; Hamasaki, M.; Kaneko, Y.; Iwasaki, H. Establishment of three malignant peripheral nerve sheath tumor cell lines, FU-SFT8611, 8710 and 9817: Conventional and molecular cytogenetic characterization. Int. J. Oncol. 2006, 29, 1421–1428. [Google Scholar] [CrossRef]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Neuro, A.O. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008, 10, 946–957. [Google Scholar] [CrossRef]
- Imaizumi, S.; Motoyama, T.; Ogose, A.; Hotta, T.; Takahashi, H.E. Characterization and chemosensitivity of two human malignant peripheral nerve sheath tumour cell lines derived from a patient with neurofibromatosis type 1. Virchows Arch. 1998, 433, 435–441. [Google Scholar] [CrossRef]
- Subramanian, S.; Thayanithy, V.; West, R.B.; Lee, C.-H.; Beck, A.H.; Zhu, S.; Downs-Kelly, E.; Montgomery, K.; Goldblum, J.R.; Hogendoorn, P.C.; et al. Genome-wide transcriptome analyses reveal p53 inactivation mediated loss of miR-34a expression in malignant peripheral nerve sheath tumours. J. Pathol. 2010, 220, 58–70. [Google Scholar] [CrossRef]
- Lopez, G.; Torres, K.; Liu, J.; Hernandez, B.; Young, E.; Belousov, R.; Bolshakov, S.; Lazar, A.J.; Slopis, J.M.; McCutcheon, I.E.; et al. Autophagic Survival in Resistance to Histone Deacetylase Inhibitors: Novel Strategies to Treat Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2011, 71, 185–196. [Google Scholar] [CrossRef]
- Spyra, M.; Kluwe, L.; Hagel, C.; Nguyen, R.; Panse, J.; Kurtz, A.; Mautner, V.-F.; Rabkin, S.D.; Demestre, M. Cancer Stem Cell-Like Cells Derived from Malignant Peripheral Nerve Sheath Tumors. PLoS ONE 2011, 6, e21099. [Google Scholar] [CrossRef]
- Badache, A.; De Vries, G.H. Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J. Cell. Physiol. 1998, 177, 334–342. [Google Scholar] [CrossRef]
- Sonobe, H.; Takeuchi, T.; Furihata, M.; Taguchi, T.; Kawai, A.; Ohjimi, Y.; Iwasaki, H.; Kaneko, Y.; Ohtsuki, Y. A new human malignant peripheral nerve sheath tumour-cell line, HS-sch-2, harbouring p53 point mutation. Int. J. Oncol. 2000, 17, 347–352. [Google Scholar] [CrossRef]
- Kolberg, M.; Bruun, J.; Murumägi, A.; Mpindi, J.P.; Bergsland, C.H.; Høland, M.; Eilertsen, I.A.; Danielsen, S.A.; Kallioniemi, O.; Lothe, R.A. Drug sensitivity and resistance testing identifies PLK1 inhibitors and gemcitabine as potent drugs for malignant peripheral nerve sheath tumors. Mol. Oncol. 2017, 11, 1156–1171. [Google Scholar] [CrossRef]
- Schoffski, P.; Van Renterghem, B.; Cornillie, J.; Wang, Y.; Gebreyohannes, Y.K.; Lee, C.-J.; Wellens, J.; Vanleeuw, U.; Nysen, M.; Hompes, D.; et al. XenoSarc: A comprehensive platform of patient-derived xenograft (PDX) models of soft tissue sarcoma (STS) for early drug testing. J. Glob. Oncol. 2019, 5, 37. [Google Scholar] [CrossRef]
- Castellsague, J.; Gel, B.; Fernandez-Rodriguez, J.; Llatjos, R.; Blanco, I.; Benavente, Y.; Perez-Sidelnikova, D.; Garcia-del Muro, J.; Vinals, J.M.; Vidal, A.; et al. Comprehensive establishment and characterization of orthoxenograft mouse models of malignant peripheral nerve sheath tumors for personalized medicine. EMBO Mol. Med. 2015, 7, 608–627. [Google Scholar] [CrossRef]
- Brossier, N.M.; Carroll, S.L. Genetically engineered mouse models shed new light on the pathogenesis of neurofibromatosis type I-related neoplasms of the peripheral nervous system. Brain Res. Bull. 2012, 88, 58–71. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Cell Line | Sex | Synonyms | Origin | PRC2 Status | Ref. |
|---|---|---|---|---|---|
| T265 | / | T265-2c; T265-2C; T265p21 | NF1 | Loss [62,143] | [144,145] |
| 90-8 | / | MPNST 90-8TL; 90-8TL; NF90-8; NF190-8 | NF1 | Loss [19] | [146] |
| ST88-3 | M | 88-3; NF188-3 | NF1 | Unknown | [147] |
| ST88-14 | M | ST88.14; ST 88-14; ST-8814; ST8814; 88-14; NF188-14 | NF1 | Loss [18,19] | [147] |
| sNF02.2 | M | sNF02-2 | NF1 | WT [148] | [149] |
| sNF10.1 | / | NF1 | Loss [150] | [150] | |
| sNF94.3 | F | NF1 | Loss [150] | [151] | |
| sNF96.2 | M | SNF96.2; sNF96-2 | NF1 | Loss [19,148] | [152] |
| S462 | / | NF1 | Loss [19,62] | [153] | |
| S462.TY | / | S462-TY; S462TY | NF1 | Unknown | [154] |
| S520 | / | NF1 | Unknown | [153] | |
| S805 | / | NF1 | Unknown | [155] | |
| FMS-1 | F | NF1 | Unknown | [156] | |
| FU-SFT8710 | F | NF1 | Unknown | [157] | |
| NFS-1 | / | NF1 | Unknown | [158] | |
| NMS-2 | M | NF1 | Unknown | [159] | |
| NMS-2PC | M | NF1 | Unknown | [159] | |
| MPNST-14 | M | NF1 | Unknown | [160] | |
| MPNST642 | M | NF1 | Unknown | [161] | |
| 1507.2 | / | S1507-2 | NF1 | Unknown | [153,162] |
| STS-26T | / | STS26T; STS26 | Sporadic | WT [62,143] | [163] |
| MPNST-724 | / | MPNST724 | Sporadic | WT [18] | [160] |
| HS-Sch-2 | F | Sporadic | Unknown | [164] | |
| HS-PSS | M | Sporadic | Unknown | [165] | |
| YST-1 | F | Sporadic | Unknown | [165] | |
| FU-SFT9817 | F | Sporadic | Unknown | [157] | |
| FU-SFT8611 | M | Sporadic | Unknown | [157] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Murray, B.; Mo, G.; Shern, J.F. The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor. Genes 2020, 11, 287. https://doi.org/10.3390/genes11030287
Zhang X, Murray B, Mo G, Shern JF. The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor. Genes. 2020; 11(3):287. https://doi.org/10.3390/genes11030287
Chicago/Turabian StyleZhang, Xiyuan, Béga Murray, George Mo, and Jack F. Shern. 2020. "The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor" Genes 11, no. 3: 287. https://doi.org/10.3390/genes11030287
APA StyleZhang, X., Murray, B., Mo, G., & Shern, J. F. (2020). The Role of Polycomb Repressive Complex in Malignant Peripheral Nerve Sheath Tumor. Genes, 11(3), 287. https://doi.org/10.3390/genes11030287