Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1

 ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials Implementation and Data and Code Availability
2.2. Sequencing Data Collection and Processing
2.3. Latent Variable Calculation and Selection
2.4. Generation of Ensemble of Random Forests for Feature Selection
2.4.1. Algorithm Implementation
2.4.2. Feature Selection
2.5. Immune Subtype Prediction
2.6. MetaVIPER
2.7. VIPER Correlation Clustering and Drug Enrichment Analysis
3. Results
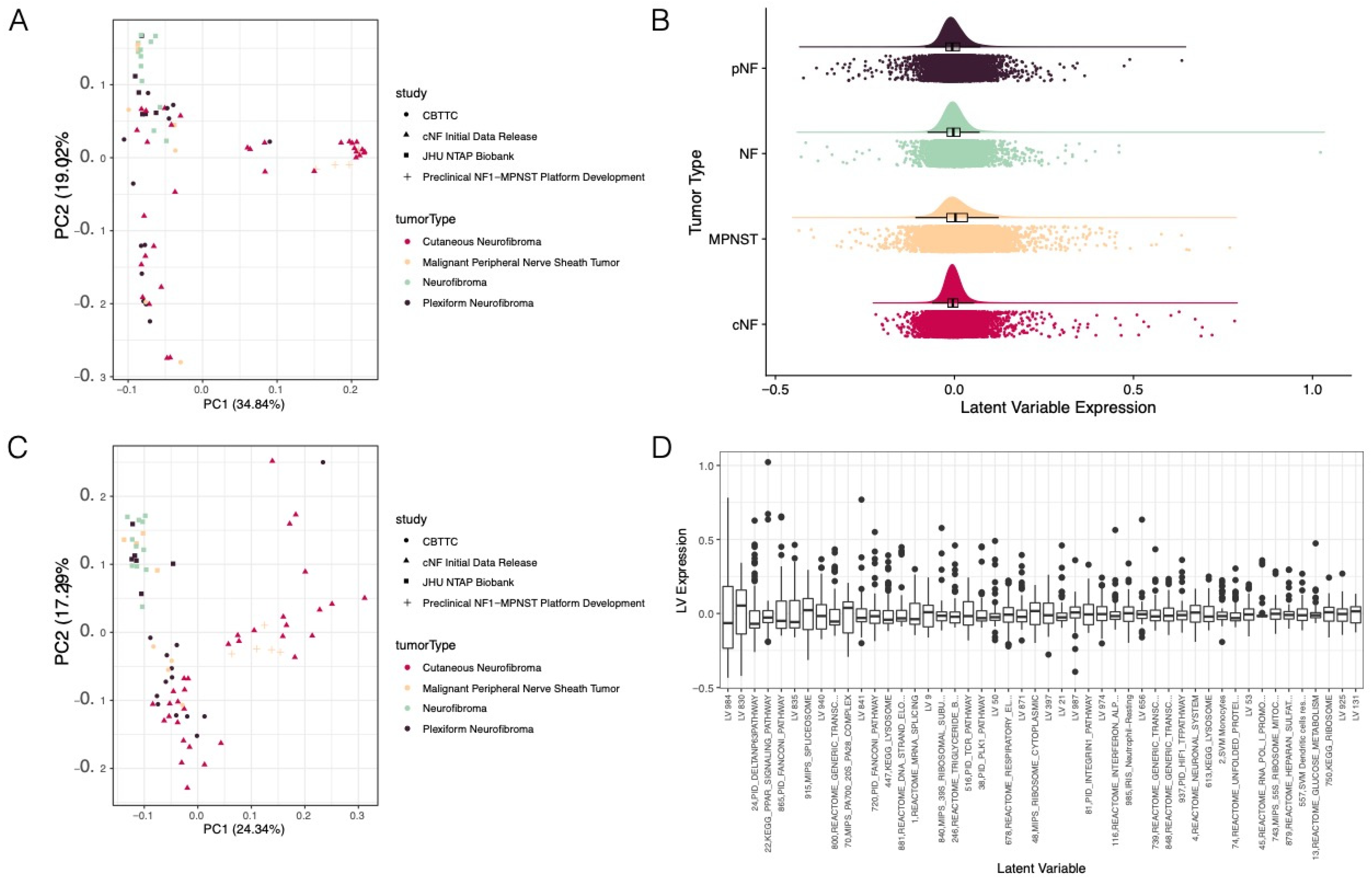
3.1. Pan-NF Transcriptomic Analysis Identified Most Variable Latent Variables in NF1
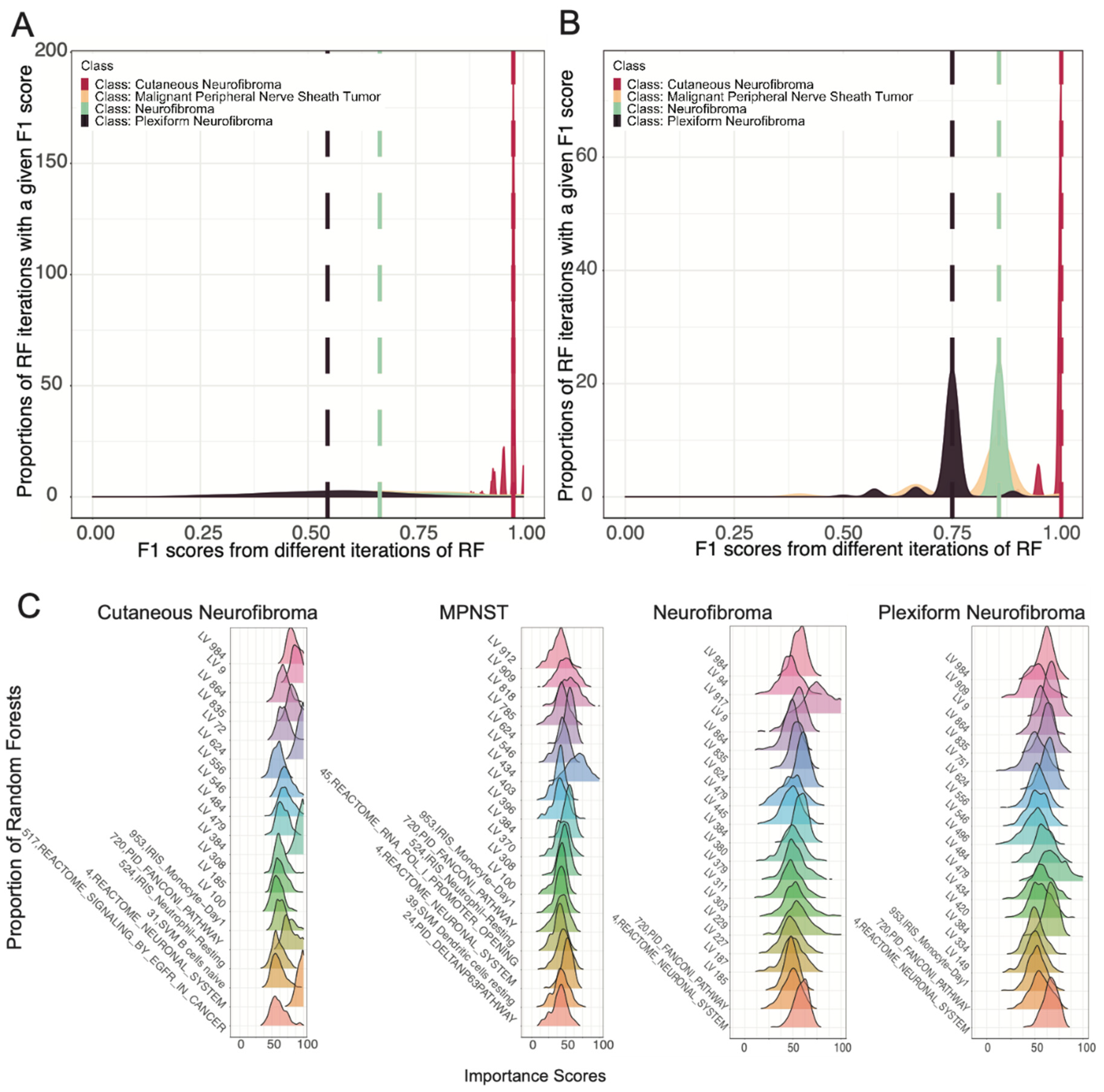
3.2. Ensemble of Random Forests Identified Latent Variables That Robustly Describe Individual Tumor Types
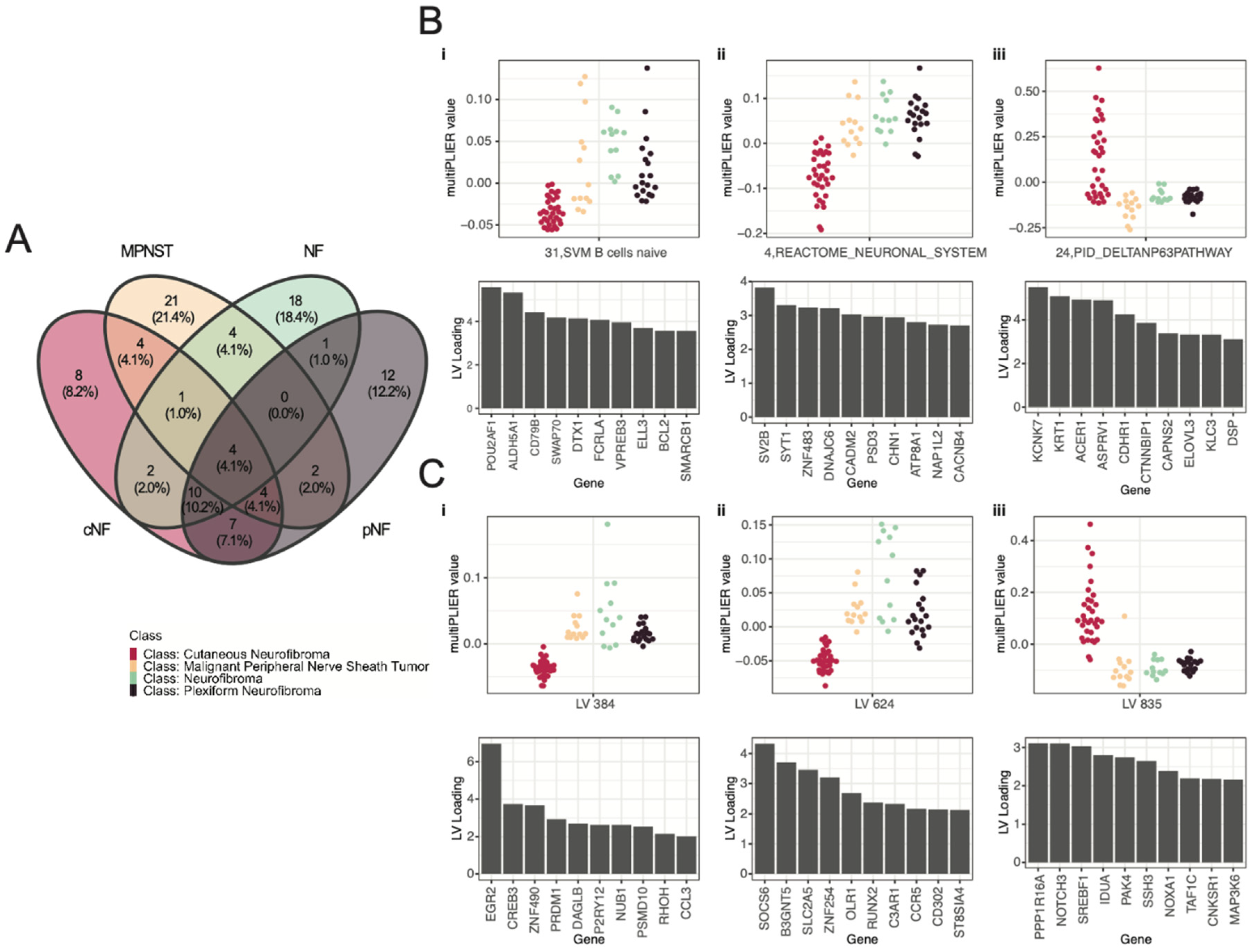
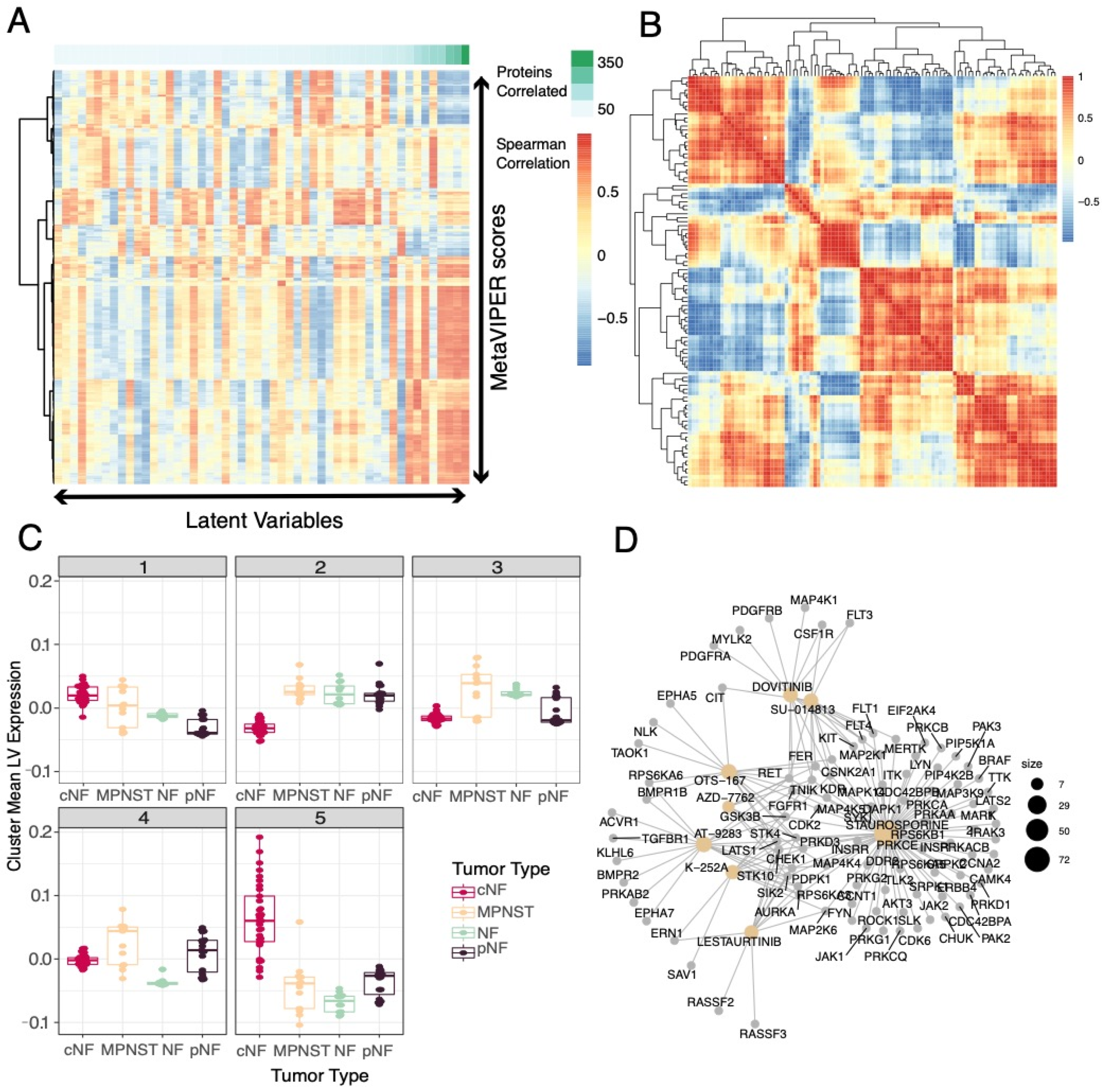
3.3. Selected Latent Variables Represented Distinct Biology of Nerve Sheath Tumor Types
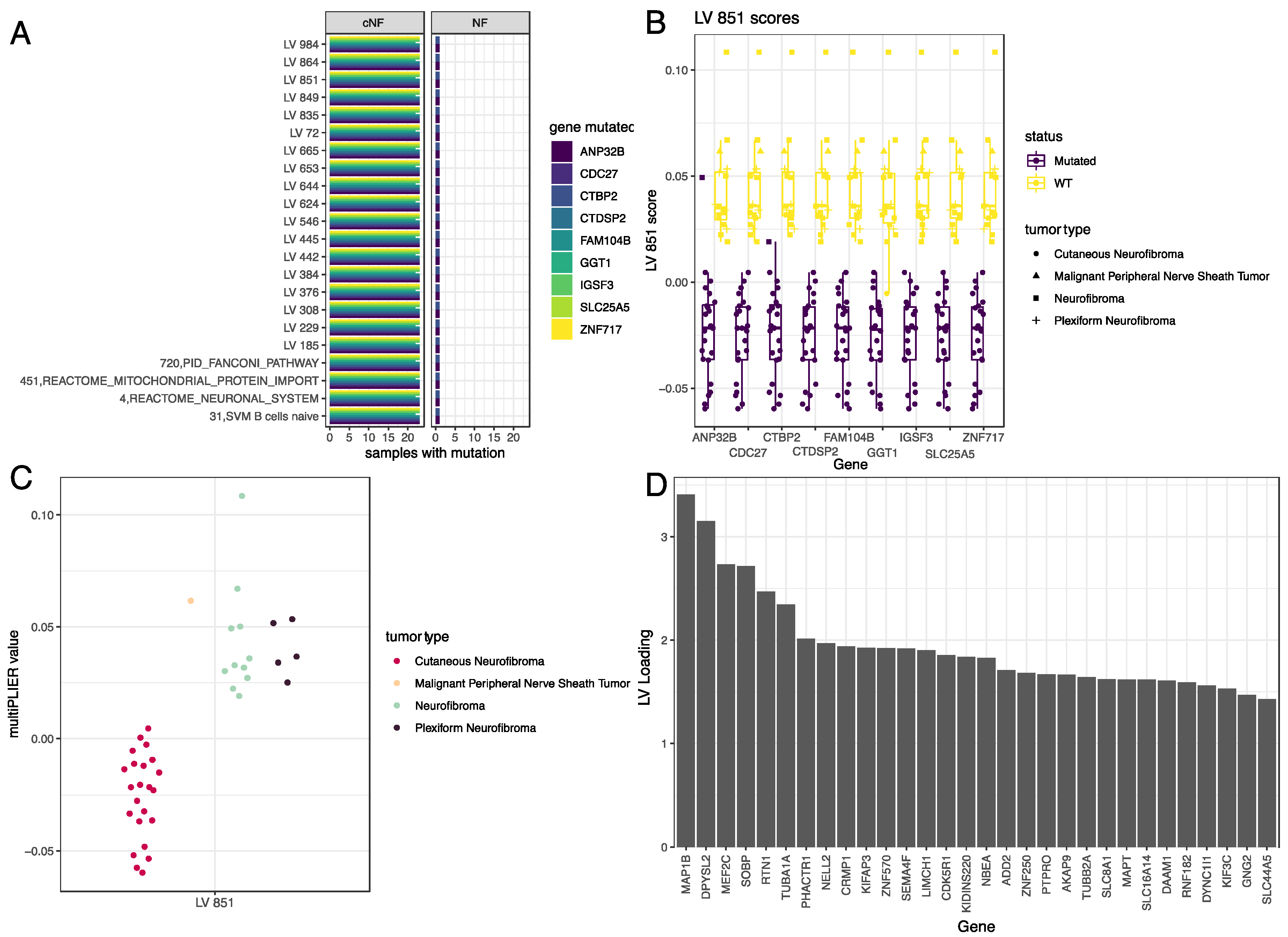
3.4. LV Scores May Be Attributed to Specific Gene Variants for Specific Tumor Types
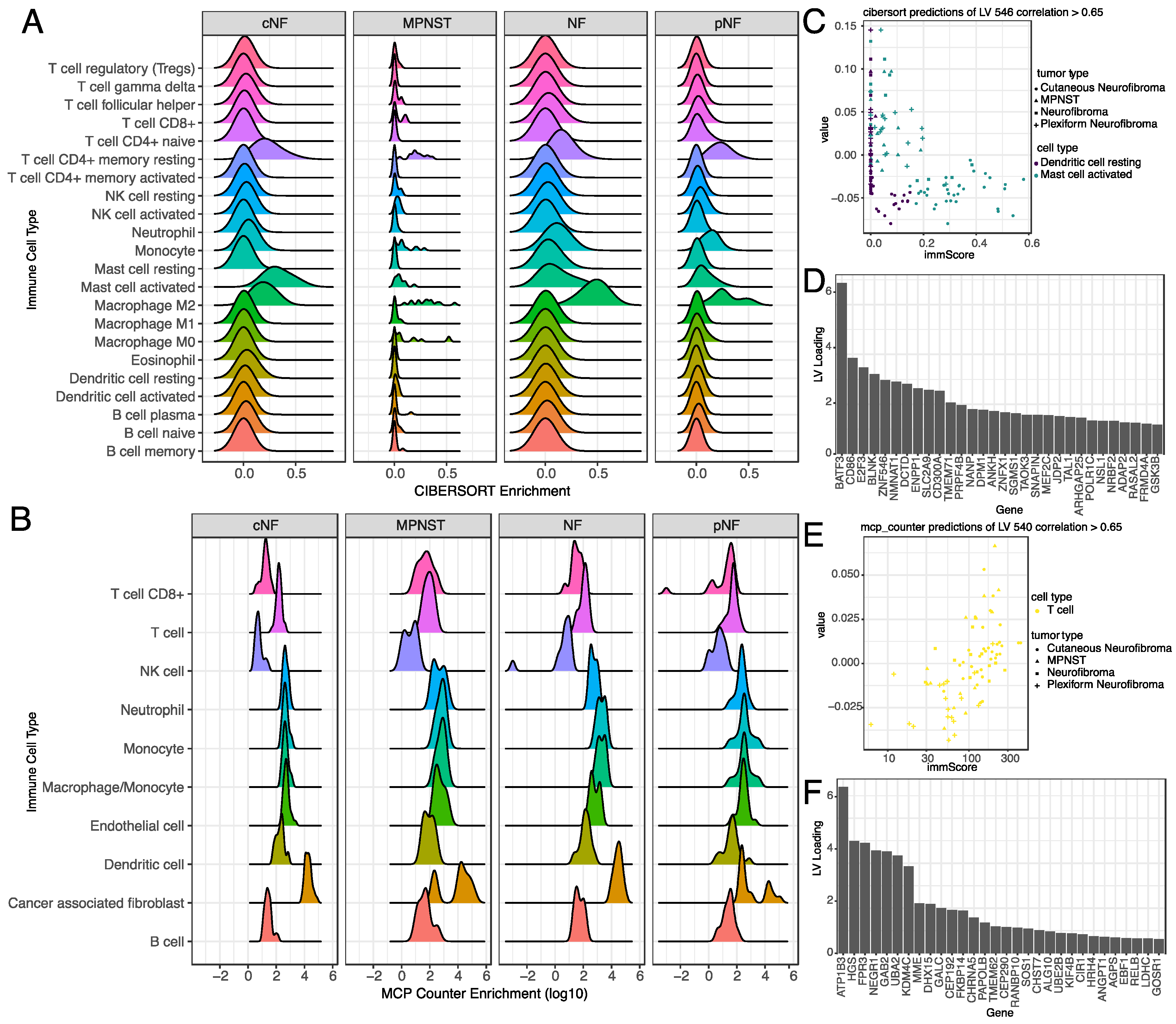
3.5. Selected Latent Variables Represented Specific Immune Cell Types in the Tumor Microenvironment
3.6. Selected Latent Variables Captured Protein Regulatory Networks in NF1 Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, J.M. Epidemiology of neurofibromatosis type 1. Am. J. Med. Genet. 1999, 89, 1–6. [Google Scholar] [CrossRef]
- Evans, D.G.; Howard, E.; Giblin, C.; Clancy, T.; Spencer, H.; Huson, S.M.; Lalloo, F. Birth incidence and prevalence of tumor-prone syndromes: Estimates from a UK family genetic register service. Am. J. Med. Genet. A 2010, 152A, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Allaway, R.J.; Gosline, S.J.C.; La Rosa, S.; Knight, P.; Bakker, A.; Guinney, J.; Le, L.Q. Cutaneous neurofibromas in the genomics era: Current understanding and open questions. Br. J. Cancer 2018, 118, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, S.R.; Bredella, M.A.; Cai, W.; Kassarjian, A.; Harris, G.J.; Esparza, S.; Merker, V.L.; Munn, L.L.; Muzikansky, A.; Askenazi, M.; et al. Quantitative Assessment of Whole-Body Tumor Burden in Adult Patients with Neurofibromatosis. PLoS ONE 2012, 7, e35711. [Google Scholar] [CrossRef]
- Stucky, C.-C.H.; Johnson, K.N.; Gray, R.J.; Pockaj, B.A.; Ocal, I.T.; Rose, P.S.; Wasif, N. Malignant Peripheral Nerve Sheath Tumors (MPNST): The Mayo Clinic Experience. Ann. Surg. Oncol. 2012, 19, 878–885. [Google Scholar] [CrossRef]
- Yuan, Z.; Xu, L.; Zhao, Z.; Xu, S.; Zhang, X.; Liu, T.; Zhang, S.; Yu, S. Clinicopathological features and prognosis of malignant peripheral nerve sheath tumor: A retrospective study of 159 cases from 1999 to 2016. Oncotarget 2017, 8, 104785–104795. [Google Scholar] [CrossRef]
- Peacock, J.D.; Pridgeon, M.G.; Tovar, E.A.; Essenburg, C.J.; Bowman, M.; Madaj, Z.; Koeman, J.; Boguslawski, E.A.; Grit, J.; Dodd, R.D.; et al. Genomic Status of MET Potentiates Sensitivity to MET and MEK Inhibition in NF1-Related Malignant Peripheral Nerve Sheath Tumors. Cancer Res. 2018, 78, 3672–3687. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef]
- D’Angelo, F.; Ceccarelli, M.; Tala; Garofano, L.; Zhang, J.; Frattini, V.; Caruso, F.P.; Lewis, G.; Alfaro, K.D.; Bauchet, L.; et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 2019, 25, 176–187. [Google Scholar]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.V.; Eaves, D.; Jessen, W.J.; Rizvi, T.A.; Ecsedy, J.A.; Qian, M.G.; Aronow, B.J.; Perentesis, J.P.; Serra, E.; Cripe, T.P.; et al. Ras-Driven Transcriptome Analysis Identifies Aurora Kinase A as a Potential Malignant Peripheral Nerve Sheath Tumor Therapeutic Target. Clin. Cancer Res. 2012, 18, 5020–5030. [Google Scholar] [CrossRef] [PubMed]
- Jessen, W.J.; Miller, S.J.; Jousma, E.; Wu, J.; Rizvi, T.A.; Brundage, M.E.; Eaves, D.; Widemann, B.; Kim, M.-O.; Dombi, E.; et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J. Clin. Investig. 2013, 123, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Komurov, K.; Fletcher, J.S.; Jousma, E.; Cancelas, J.A.; Wu, J.; Ratner, N. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci. Rep. 2017, 7, 43315. [Google Scholar] [CrossRef]
- Pemov, A.; Li, H.; Patidar, R.; Hansen, N.F.; Sindiri, S.; Hartley, S.W.; Wei, J.S.; Elkahloun, A.; Chandrasekharappa, S.C.; Boland, J.F.; et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene 2017, 36, 3168–3177. [Google Scholar] [CrossRef]
- Pemov, A.; Hansen, N.F.; Sindiri, S.; Patidar, R.; Higham, C.S.; Dombi, E.; Miettinen, M.M.; Fetsch, P.; Brems, H.; Chandrasekharappa, S.; et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define pre-malignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro-Oncology 2019, 21, 981–992. [Google Scholar] [CrossRef]
- Thomas, L.; Kluwe, L.; Chuzhanova, N.; Mautner, V.; Upadhyaya, M. Analysis of NF1 somatic mutations in cutaneous neurofibromas from patients with high tumor burden. Neurogenetics 2010, 11, 391–400. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- International network of cancer genome projects. Nature 2010, 464, 993–998. [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Ali, M.; Aittokallio, T. Machine learning and feature selection for drug response prediction in precision oncology applications. Biophys. Rev. 2019, 11, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.C.; Heiser, L.M.; Georgii, E.; Gönen, M.; Menden, M.P.; Wang, N.J.; Bansal, M.; Ammad-ud-din, M.; Hintsanen, P.; Khan, S.A.; et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat. Biotechnol. 2014, 32, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Sanchez-Vega, F.; La, K.; Armenia, J.; Chatila, W.K.; Luna, A.; Sander, C.; Cherniack, A.D.; Mina, M.; Ciriello, G.; et al. Machine Learning Detects Pan-cancer Ras Pathway Activation in The Cancer Genome Atlas. Cell Rep. 2018, 23, 172–180.e3. [Google Scholar] [CrossRef] [PubMed]
- Allaway, R.J.; La Rosa, S.; Verma, S.; Mangravite, L.; Guinney, J.; Blakeley, J.; Bakker, A.; Gosline, S.J.C. Engaging a community to enable disease-centric data sharing with the NF Data Portal. Sci. Data 2019, 6, 319. [Google Scholar] [CrossRef] [PubMed]
- Collado-Torres, L.; Nellore, A.; Kammers, K.; Ellis, S.E.; Taub, M.A.; Hansen, K.D.; Jaffe, A.E.; Langmead, B.; Leek, J.T. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 2017, 35, 319–321. [Google Scholar] [CrossRef]
- Mao, W.; Zaslavsky, E.; Hartmann, B.M.; Sealfon, S.C.; Chikina, M. Pathway-level information extractor (PLIER) for gene expression data. Nat. Methods 2019, 16, 607–610. [Google Scholar] [CrossRef]
- Taroni, J.N.; Grayson, P.C.; Hu, Q.; Eddy, S.; Kretzler, M.; Merkel, P.A.; Greene, C.S. MultiPLIER: A Transfer Learning Framework for Transcriptomics Reveals Systemic Features of Rare Disease. Cell Syst. 2019, 8, 380–394. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautès-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Sturm, G.; Finotello, F.; Petitprez, F.; Zhang, J.D.; Baumbach, J.; Fridman, W.H.; List, M.; Aneichyk, T. Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019, 35, i436–i445. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Douglass, E.F.; Sonabend, A.M.; Mela, A.; Bose, S.; Gonzalez, C.; Canoll, P.D.; Sims, P.A.; Alvarez, M.J.; Califano, A. Quantitative assessment of protein activity in orphan tissues and single cells using the metaVIPER algorithm. Nat. Commun. 2018, 9, 1471. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Hoff, B.; Ladia, K. Synapser: R Language Bindings for Synapse API. 2019. Available online: https://r-docs.synapse.org/ (accessed on 19 February 2020).
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2016, 4, 1521. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon: Fast and bias-aware quantification of transcript expression using dual-phase inference. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Pollard, K.; Banerjee, J.; Doan, X.; Wang, J.; Guo, X.; Allaway, R.; Langmead, S.; Slobogean, B.; Meyer, C.F.; Loeb, D.M.; et al. A clinically and genomically annotated nerve sheath tumor biospecimen repository. bioRxiv 2019. [Google Scholar] [CrossRef]
- Gosline, S.J.C.; Weinberg, H.; Knight, P.; Yu, T.; Guo, X.; Prasad, N.; Jones, A.; Shrestha, S.; Boone, B.; Levy, S.E.; et al. A high-throughput molecular data resource for cutaneous neurofibromas. Sci. Data 2017, 4, 170045. [Google Scholar] [CrossRef]
- Ijaz, H.; Koptyra, M.; Gaonkar, K.S.; Rokita, J.L.; Baubet, V.P.; Tauhid, L.; Zhu, Y.; Brown, M.; Lopez, G.; Zhang, B.; et al. Pediatric high-grade glioma resources from the Children’s Brain Tumor Tissue Consortium. Neuro-Oncology 2020, 22, 163–165. [Google Scholar] [CrossRef]
- Carlson, M. org.Hs.eg.db: Genome Wide Annotation for Human; R Package Version 3.8.2; R Development Core Team: Vienna, Austria, 2019. [Google Scholar]
- MultiPLIER Fileset 2019. Available online: https://doi.org/10.6084/m9.figshare.6982919.v2 (accessed on 19 February 2020).
- Collado-Torres, L.; Nellore, A.; Jaffe, A.E.; Taub, M.A.; Kammers, K.; Ellis, S.E.; Hansen, K.D.; Langmead, B.; Leek, J.T. Recount: Explore and Download Data from the Recount Project; Bioconductor Version: Release (3.10). 2019. Available online: https://rdrr.io/bioc/recount/ (accessed on 19 February 2020).
- Tan, J.; Huyck, M.; Hu, D.; Zelaya, R.A.; Hogan, D.A.; Greene, C.S. ADAGE signature analysis: Differential expression analysis with data-defined gene sets. BMC Bioinform. 2017, 18, 512. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 5. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Giorgi, F.M. aracne.networks: ARACNe-inferred gene networks from TCGA tumor datasets. R Package Vers. 2018. [Google Scholar]
- Kolde, R. pheatmap: Pretty Heatmaps. R Package Vers. 2019, 61, 617. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Allaway, R.J.; La Rosa, S.; Guinney, J.; Gosline, S.J.C. Probing the chemical-biological relationship space with the Drug Target Explorer. J. Cheminform. 2018, 10, 41. [Google Scholar] [CrossRef]
- Yu, G. enrichplot: Visualization of Functional Enrichment Result. R Package Vers. 2019, 112. [Google Scholar]
- Laks, D.R.; Oses-Prieto, J.A.; Alvarado, A.G.; Nakashima, J.; Chand, S.; Azzam, D.B.; Gholkar, A.A.; Sperry, J.; Ludwig, K.; Condro, M.C.; et al. A molecular cascade modulates MAP1B and confers resistance to mTOR inhibition in human glioblastoma. Neuro-Oncology 2018, 20, 764–775. [Google Scholar] [CrossRef]
- Margolin, A.A.; Nemenman, I.; Basso, K.; Wiggins, C.; Stolovitzky, G.; Dalla Favera, R.; Califano, A. ARACNE: An algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinform. 2006, 7 (Suppl. 1), S7. [Google Scholar] [CrossRef]
- Sevilla, T.; Sivera, R.; Martínez-Rubio, D.; Lupo, V.; Chumillas, M.J.; Calpena, E.; Dopazo, J.; Vílchez, J.J.; Palau, F.; Espinós, C. The EGR2 gene is involved in axonal Charcot−Marie−Tooth disease. Eur. J. Neurol. 2015, 22, 1548–1555. [Google Scholar] [CrossRef]
- Warner, L.E.; Mancias, P.; Butler, I.J.; McDonald, C.M.; Keppen, L.; Koob, K.G.; Lupski, J.R. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat. Genet. 1998, 18, 382–384. [Google Scholar] [CrossRef]
- Roos, K.L.; Pascuzzi, R.M.; Dunn, D.W. Neurofibromatosis, Charcot-Marie-Tooth disease, or both? Neurofibromatosis 1989, 2, 238–243. [Google Scholar]
- Lancaster, E.; Elman, L.B.; Scherer, S.S. A patient with Neurofibromatosis type 1 and Charcot-Marie-Tooth Disease type 1B. Muscle Nerve 2010, 41, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Lupski, J.R.; Pentao, L.; Williams, L.L.; Patel, P.I. Stable inheritance of the CMT1A DNA duplication in two patients with CMT1 and NF1. Am. J. Med. Genet. 1993, 45, 92–96. [Google Scholar] [CrossRef]
- Lin, H.-P.; Oksuz, I.; Svaren, J.; Awatramani, R. Egr2-dependent microRNA-138 is dispensable for peripheral nerve myelination. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and Dermal Neurofibromas and Pigmentation Are Caused by Nf1 Loss in Desert Hedgehog-Expressing Cells. Cancer Cell 2008, 13, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Kabir, N.N.; Sun, J.; Rönnstrand, L.; Kazi, J.U. SOCS6 is a selective suppressor of receptor tyrosine kinase signaling. Tumor Biol. 2014, 35, 10581–10589. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Wang, W.; Su, J.; Zhang, Y.; Luan, B.; Rao, H.; Cheng, T.; Zhang, W.; Xiao, S.; Zhang, M.; et al. SOCS6 Functions as a Tumor Suppressor by Inducing Apoptosis and Inhibiting Angiogenesis in Human Prostate Cancer. Available online: http://www.eurekaselect.com/158762/article (accessed on 10 February 2020).
- Hall, A.; Choi, K.; Liu, W.; Rose, J.; Zhao, C.; Yu, Y.; Na, Y.; Cai, Y.; Coover, R.A.; Lin, Y.; et al. RUNX represses Pmp22 to drive neurofibromagenesis. Sci. Adv. 2019, 5, eaau8389. [Google Scholar] [CrossRef]
- Veremeyko, T.; Yung, A.W.Y.; Anthony, D.C.; Strekalova, T.; Ponomarev, E.D. Early Growth Response Gene-2 Is Essential for M1 and M2 Macrophage Activation and Plasticity by Modulation of the Transcription Factor CEBPβ. Front. Immunol. 2018, 9, 2515. [Google Scholar] [CrossRef]
- Okamura, T.; Yamamoto, K.; Fujio, K. Early Growth Response Gene 2-Expressing CD4+LAG3+ Regulatory T Cells: The Therapeutic Potential for Treating Autoimmune Diseases. Front. Immunol. 2018, 9, 340. [Google Scholar] [CrossRef]
- Hamdan, F.H.; Johnsen, S.A. DeltaNp63-dependent super enhancers define molecular identity in pancreatic cancer by an interconnected transcription factor network. Proc. Natl. Acad. Sci. USA 2018, 115, E12343–E12352. [Google Scholar] [CrossRef]
- Cancino, G.I.; Yiu, A.P.; Fatt, M.P.; Dugani, C.B.; Flores, E.R.; Frankland, P.W.; Josselyn, S.A.; Miller, F.D.; Kaplan, D.R. p63 Regulates Adult Neural Precursor and Newly Born Neuron Survival to Control Hippocampal-Dependent Behavior. J. Neurosci. 2013, 33, 12569–12585. [Google Scholar] [CrossRef]
- Packard, A.; Schnittke, N.; Romano, R.-A.; Sinha, S.; Schwob, J.E. ΔNp63 Regulates Stem Cell Dynamics in the Mammalian Olfactory Epithelium. J. Neurosci. 2011, 31, 8748–8759. [Google Scholar] [CrossRef] [PubMed]
- Carr, N.J.; Warren, A.Y. Mast cell numbers in melanocytic naevi and cutaneous neurofibromas. J. Clin. Pathol. 1993, 46, 86–87. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhu, Y.; Ghosh, P.; Charnay, P.; Burns, D.K.; Parada, L.F. Neurofibromas in NF1: Schwann Cell Origin and Role of Tumor Environment. Science 2002, 296, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, P. Mast cells in benign nerve sheath tumours. J. Pathol. 1976, 119, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Tucker, T.; Riccardi, V.M.; Sutcliffe, M.; Vielkind, J.; Wechsler, J.; Wolkenstein, P.; Friedman, J.M. Different patterns of mast cells distinguish diffuse from encapsulated neurofibromas in patients with neurofibromatosis 1. J. Histochem. Cytochem. 2011, 59, 584–590. [Google Scholar] [CrossRef][Green Version]
- Greggio: Les Cellules Granuleuses (Mastzellen) Dans-Google Scholar. Available online: https://scholar.google.com/scholar_lookup?journal=Arch.+Med.+Exp.&title=Les+cellules+granuleuses+(Mastzellen)+dans+les+tissus+normaux+et+dans+certaines+maladies+chirurgicales&author=H+Greggio&volume=23&publication_year=1911&pages=323-375& (accessed on 13 January 2020).
- Chen, Z.; Mo, J.; Brosseau, J.-P.; Shipman, T.; Wang, Y.; Liao, C.-P.; Cooper, J.M.; Allaway, R.J.; Gosline, S.J.C.; Guinney, J.; et al. Spatiotemporal Loss of NF1 in Schwann Cell Lineage Leads to Different Types of Cutaneous Neurofibroma Susceptible to Modification by the Hippo Pathway. Cancer Discov. 2019, 9, 114–129. [Google Scholar] [CrossRef]
- Nürnberger, M.; Moll, I. Semiquantitative aspects of mast cells in normal skin and in neurofibromas of neurofibromatosis types 1 and 5. Dermatology 1994, 188, 296–299. [Google Scholar] [CrossRef]
- Brosseau, J.-P.; Liao, C.-P.; Wang, Y.; Ramani, V.; Vandergriff, T.; Lee, M.; Patel, A.; Ariizumi, K.; Le, L.Q. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Farschtschi, S.; Park, S.-J.; Sawitzki, B.; Oh, S.-J.; Kluwe, L.; Mautner, V.F.; Kurtz, A. Effector T cell subclasses associate with tumor burden in neurofibromatosis type 1 patients. Cancer Immunol. Immunother. 2016, 65, 1113–1121. [Google Scholar] [CrossRef]
- Kahen, E.J.; Brohl, A.; Yu, D.; Welch, D.; Cubitt, C.L.; Lee, J.K.; Chen, Y.; Yoder, S.J.; Teer, J.K.; Zhang, Y.O.; et al. Neurofibromin level directs RAS pathway signaling and mediates sensitivity to targeted agents in malignant peripheral nerve sheath tumors. Oncotarget 2018, 9, 22571–22585. [Google Scholar] [CrossRef]
- Allaway, R.J.; Fischer, D.A.; de Abreu, F.B.; Gardner, T.B.; Gordon, S.R.; Barth, R.J.; Colacchio, T.A.; Wood, M.; Kacsoh, B.Z.; Bouley, S.J.; et al. Genomic characterization of patient-derived xenograft models established from fine needle aspirate biopsies of a primary pancreatic ductal adenocarcinoma and from patient-matched metastatic sites. Oncotarget 2016, 7, 17087–17102. [Google Scholar] [CrossRef] [PubMed]
- Discovery of a Small Molecule Targeting IRA2 Deletion in Budding Yeast and Neurofibromin Loss in Malignant Peripheral Nerve Sheath Tumor Cells | Molecular Cancer Therapeutics. Available online: https://mct.aacrjournals.org/content/10/9/1740.figures-only (accessed on 13 January 2020).
- Danilov, A.V.; Hu, S.; Orr, B.; Godek, K.; Mustachio, L.M.; Sekula, D.; Liu, X.; Kawakami, M.; Johnson, F.M.; Compton, D.A.; et al. Dinaciclib Induces Anaphase Catastrophe in Lung Cancer Cells via Inhibition of Cyclin-Dependent Kinases 1 and 2. Mol. Cancer Ther. 2016, 15, 2758–2766. [Google Scholar] [CrossRef] [PubMed]
- Malone, C.F.; Emerson, C.; Ingraham, R.; Barbosa, W.; Guerra, S.; Yoon, H.; Liu, L.L.; Michor, F.; Haigis, M.; Macleod, K.F.; et al. mTOR and HDAC Inhibitors Converge on the TXNIP/Thioredoxin Pathway to Cause Catastrophic Oxidative Stress and Regression of RAS-Driven Tumors. Cancer Discov. 2017, 7, 1450–1463. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Name | Synapse Project Name | Synapse Table Name | Synapse Access Team |
|---|---|---|---|
| WashU Biobank | Preclinical NF1-MPNST Platform Development (syn11638893) | WashU Biobank RNA-seq data | WUSTL MPNST PDX Data Access |
| JHU Biobank [37] | A Nerve Sheath Tumor Bank from Patients with NF1 (syn4939902) | Biobank RNASeq Data | JHU Biobank Data Access |
| cNF Patient Data [38] | Cutaneous Neurofibroma Data Resource (syn4984604) | cNF RNASeq Counts | CTF cNF Resource Data Access Group |
| CBTTC Data [39] | Children’s Brain Tumor Tissue Consortium (syn20629666) | CBTTC RNASeq Counts | CBTTC Data Access Group |
| Dataset Name | Assay | Synapse Table Name | Synapse Access Team | Synapse Project |
|---|---|---|---|---|
| JHU Biobank Exome-Seq Data | exomeSeq | Biobank ExomeSeq Data | JHU Biobank Data Access | A Nerve Sheath Tumor Bank from Patients with NF1 |
| cNF WGS Data | wholeGenomeSeq | cNF WGS Harmonized Data | CTF cNF Resource Data Access Group | Cutaneous Neurofibroma Data Resource |
| Tumor Type | Individuals | Samples | # with Genomic Variant Data |
|---|---|---|---|
| Cutaneous Neurofibroma (cNF) | 11 | 33 | 23 |
| MPNST | 13 | 13 | 1 |
| Undefined Neurofibroma (NF) | 12 | 12 | 11 |
| Plexiform Neurofibroma (pNF) | 19 | 19 | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, J.; Allaway, R.J.; Taroni, J.N.; Baker, A.; Zhang, X.; Moon, C.I.; Pratilas, C.A.; Blakeley, J.O.; Guinney, J.; Hirbe, A.; et al. Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1. Genes 2020, 11, 226. https://doi.org/10.3390/genes11020226
Banerjee J, Allaway RJ, Taroni JN, Baker A, Zhang X, Moon CI, Pratilas CA, Blakeley JO, Guinney J, Hirbe A, et al. Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1. Genes. 2020; 11(2):226. https://doi.org/10.3390/genes11020226
Chicago/Turabian StyleBanerjee, Jineta, Robert J Allaway, Jaclyn N Taroni, Aaron Baker, Xiaochun Zhang, Chang In Moon, Christine A Pratilas, Jaishri O Blakeley, Justin Guinney, Angela Hirbe, and et al. 2020. "Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1" Genes 11, no. 2: 226. https://doi.org/10.3390/genes11020226
APA StyleBanerjee, J., Allaway, R. J., Taroni, J. N., Baker, A., Zhang, X., Moon, C. I., Pratilas, C. A., Blakeley, J. O., Guinney, J., Hirbe, A., Greene, C. S., & Gosline, S. J. (2020). Integrative Analysis Identifies Candidate Tumor Microenvironment and Intracellular Signaling Pathways that Define Tumor Heterogeneity in NF1. Genes, 11(2), 226. https://doi.org/10.3390/genes11020226