Whole Genome Sequencing Indicates Heterogeneity of Hyperostotic Disorders in Dogs

 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals
2.3. Whole-Genome Resequencing
2.4. Protein and Splice Site Predictions
2.5. Availability of Data and Material
3. Results
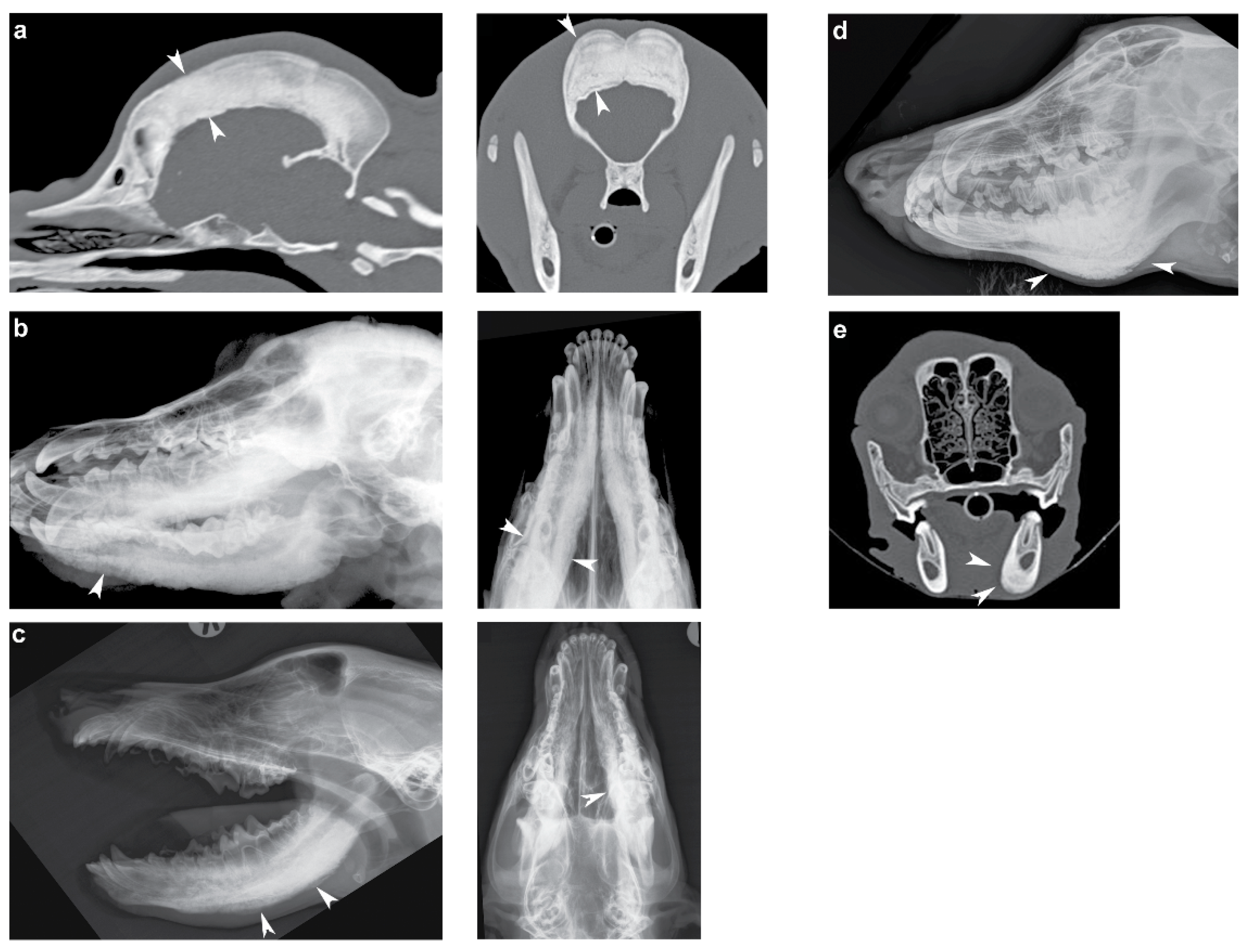
3.1. Phenotype
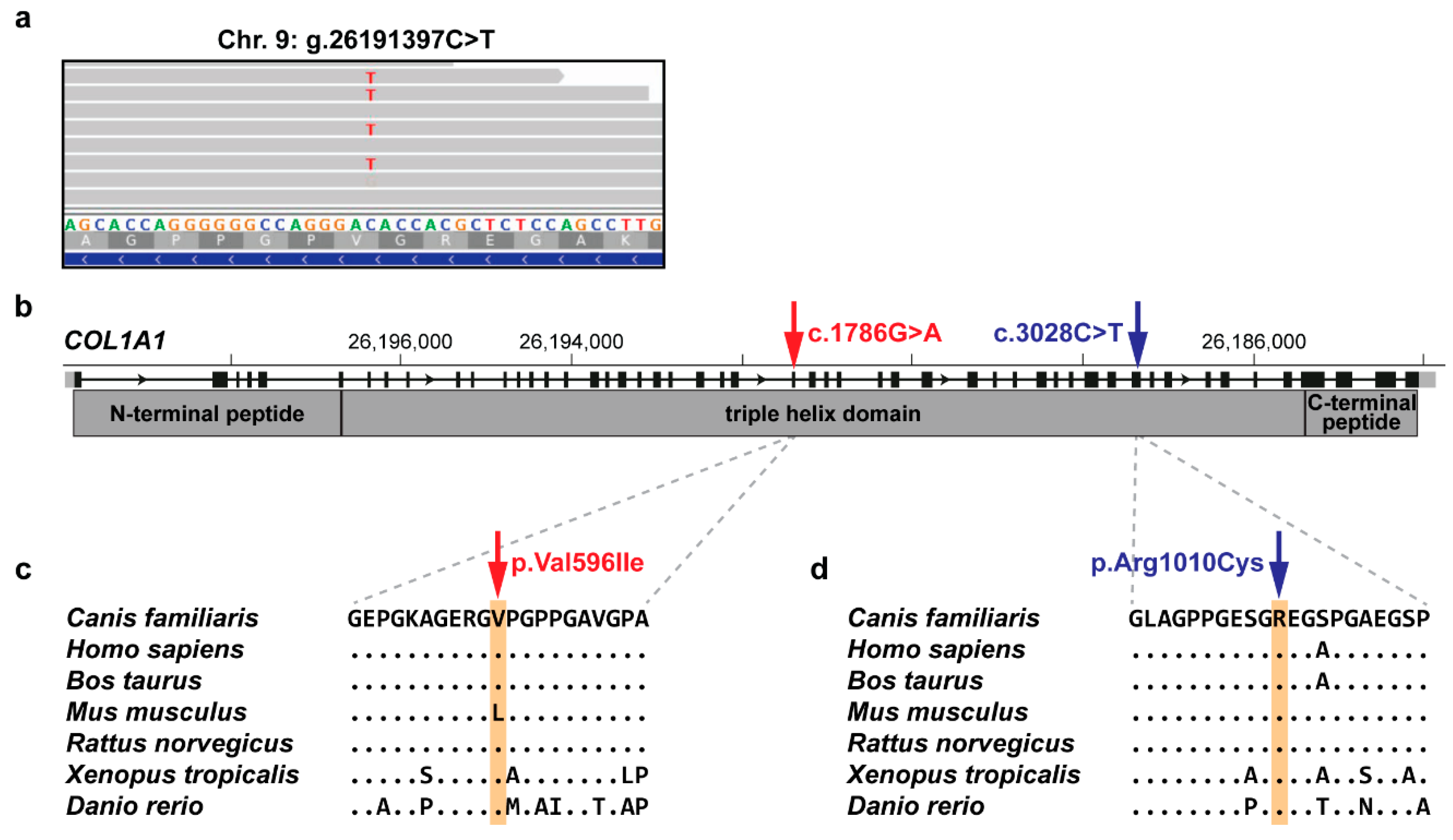
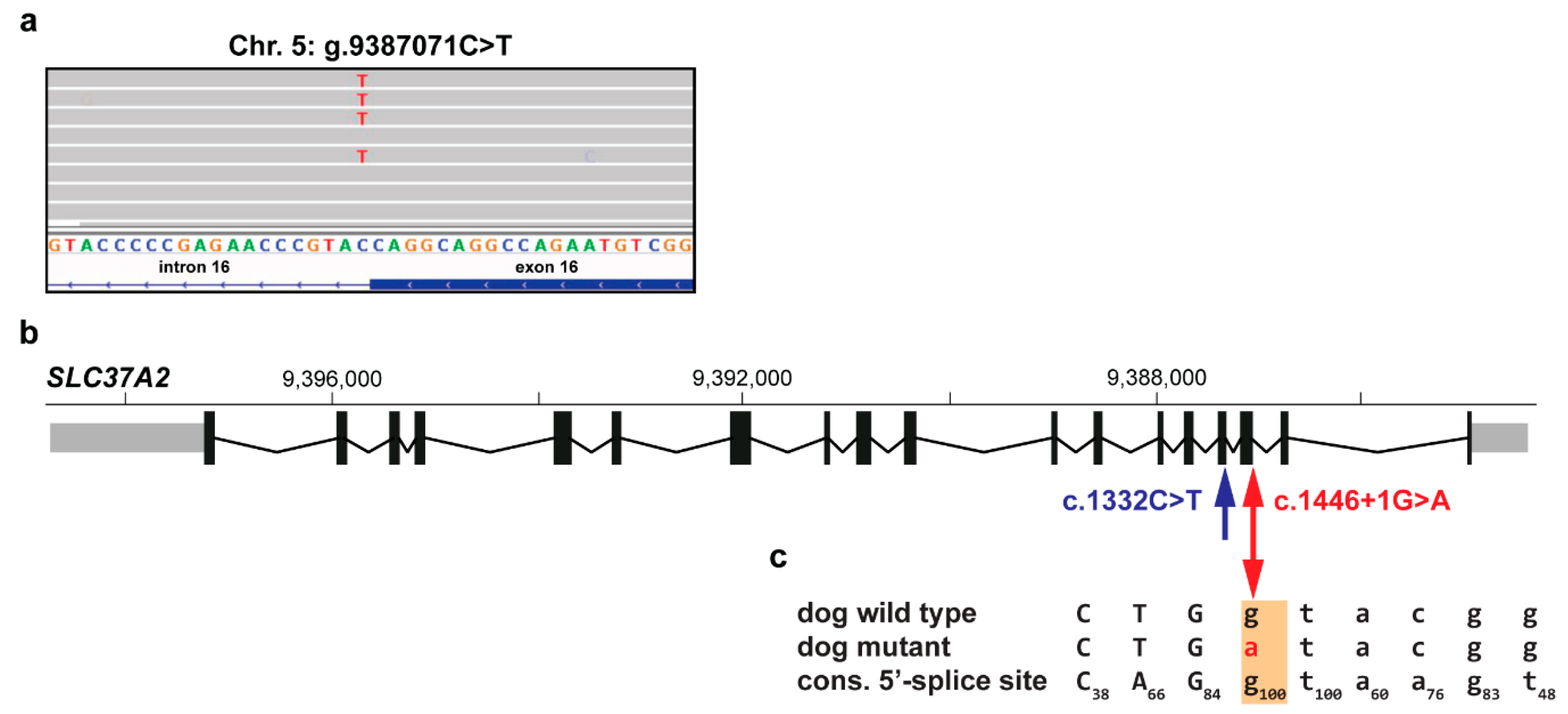
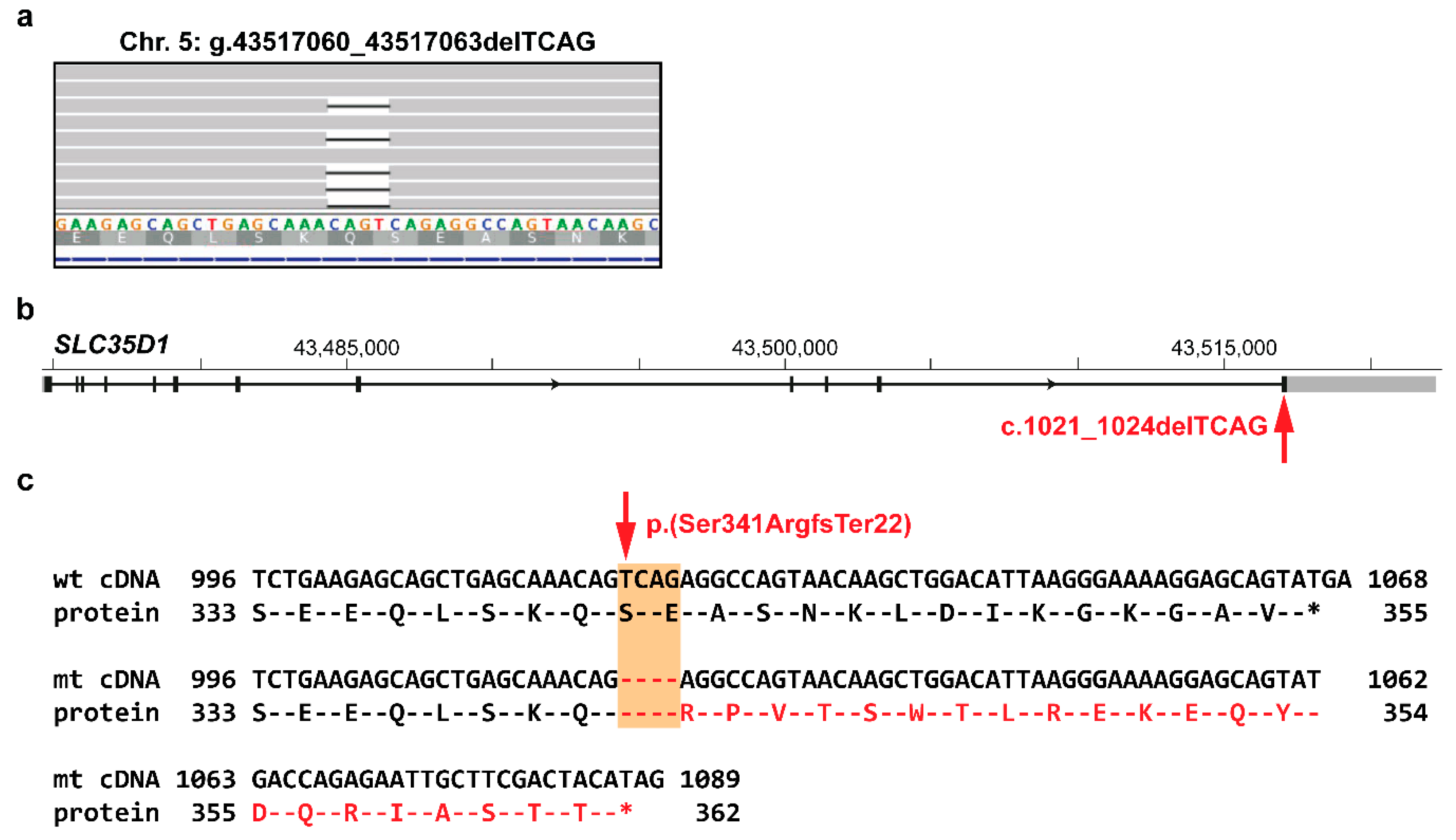
3.2. Identification of Private Candidate Variants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thornburg, L.P. Infantile cortical hyperostosis (Caffey-Silverman syndrome). Animal model: Craniomandibular osteopathy in the canine. Am. J. Pathol. 1979, 95, 575–578. [Google Scholar] [PubMed]
- Kamoun-Goldrat, A.; Le Merrer, M. Infantile Cortical Hyperostosis (Caffey Disease): A Review. J. Oral Maxillofac. Surg. 2008, 66, 2145–2150. [Google Scholar] [CrossRef] [PubMed]
- Gensure, R.C.; Cole, W.G.; Jüppner, H.; Gensure, R.C.; Mäkitie, O.; Barclay, C.; Chan, C.; Depalma, S.R.; Bastepe, M.; Abuzahra, H.; et al. A novel COL1A1 mutation in infantile cortical hyperostosis (Caffey disease) expands the spectrum of collagen-related disorders. J. Clin. Investig. 2005, 115, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Kamoun-Goldrat, A.; Martinovic, J.; Saada, J.; Sonigo-Cohen, P.; Razavi, F.; Munnich, A.; Le Merrer, M. Prenatal Cortical Hyperostosis With COL1A1 Gene Mutation. Am. J. Med. Genet. 2008, 146, 1820–1824. [Google Scholar] [CrossRef] [PubMed]
- Kitaoka, T.; Miyoshi, Y.; Namba, N.; Miura, K.; Jüppner, H.; Kubota, T.; Ohata, Y.; Fujiwara, M.; Takagi, M.; Hasegawa, T.; et al. Two Japanese familial cases of Caffey disease with and without the common COL1A1 mutation and normal bone density, and review of the literature. Eur. J. Pediatr. 2014, 173, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Merdler-rabinowicz, R.; Grinberg, A.; Jacobson, J.M.; Somekh, I.; Klein, C.; Lev, A.; Ihsan, S.; Habib, A.; Somech, R.; Simon, A.J. Fetuin-A deficiency is associated with infantile cortical hyperostosis (Caffey disease). Pediatr. Res. 2019. [Google Scholar] [CrossRef]
- Newton, C.D.; Nunamaker, D.M. Textbook of Small Animal Orthopaedics; Lippincott: Philadelphia, PA, USA, 1985; ISBN 9780397520985. [Google Scholar]
- Alexander, J.W. Selected Skeletal Dysplasias: Craniomandibular Osteopathy, Multiple Cartilaginous Exostoses, and Hypertrophic Osteodystrophy. Vet. Clin. North Am. Small Anim. Pract. 1983, 13, 55–70. [Google Scholar] [CrossRef]
- Pastor, K.F.; Boulay, J.P.; Schelling, S.H.; Carpenter, J.L. Idiopathic hyperostosis of the calvaria in five young bullmastiffs. J. Am. Anim. Hosp. Assoc. 2000, 36, 439–445. [Google Scholar] [CrossRef]
- Padgett, G.A.; Mostosky, U.V. Animal Model: The Mode of Inheritance of Craniomandibular Osteopathy in West Highland White Terrier Dogs. Am. J. Med. Genet. 1986, 13, 9–13. [Google Scholar] [CrossRef]
- Hytönen, M.K.; Arumilli, M.; Lappalainen, A.K.; Owczarek-Lipska, M.; Jagannathan, V.; Hundi, S.; Salmela, E.; Venta, P.; Sarkiala, E.; Jokinen, T.; et al. Molecular Characterization of Three Canine Models of Human Rare Bone Diseases: Caffey, van den Ende-Gupta, and Raine. PLoS Genet. 2016, 12, 1–20. [Google Scholar] [CrossRef]
- Vagt, J.; Distl, O. Complex segregation analysis of craniomandibular osteopathy in Deutsch Drahthaar dogs. Vet. J. 2018, 231, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Boycott, K.M.; Vanstone, M.R.; Bulman, D.E.; MacKenzie, A.E. Rare-disease genetics in the era of next-generation sequencing: Discovery to translation. Nat. Rev. Genet. 2013, 14, 681. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, V.; Drögemüller, C.; Leeb, T. Dog Biomedical Variant Database Consortium (DBVDC) A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and 8 wolves. Anim. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Bauer, A.; Waluk, D.P.; Galichet, A.; Timm, K.; Jagannathan, V.; Sayar, B.S.; Wiener, D.J.; Dietschi, E.; Muller, E.J.; Roosje, P.; et al. A de novo variant in the ASPRV1 gene in a dog with ichthyosis. PLoS Genet. 2017, 13, e1006651. [Google Scholar] [CrossRef]
- Hadji Rasouliha, S.; Bauer, A.; Dettwiler, M.; Welle, M.M.; Leeb, T. A frameshift variant in the EDA gene in Dachshunds with X-linked hypohidrotic ectodermal dysplasia. Anim. Genet. 2018, 49, 651–654. [Google Scholar] [CrossRef]
- Caduff, M.; Bauer, A.; Jagannathan, V.; Leeb, T. A single base deletion in the SLC45A2 gene in a Bullmastiff with oculocutaneous albinism. Anim. Genet. 2017, 48, 619–621. [Google Scholar] [CrossRef]
- Spycher, M.; Bauer, A.; Jagannathan, V.; Frizzi, M.; De Lucia, M.; Leeb, T. A frameshift variant in the COL5A1 gene in a cat with Ehlers-Danlos syndrome. Anim. Genet. 2018, 49, 641–644. [Google Scholar] [CrossRef]
- Bridavsky, M.; Kuhl, H.; Woodruff, A.; Kornak, U.; Timmermann, B.; Mages, N.; Lupianez, D.G.; Symmons, O.; Ibrahim, D.M.; 99 Lives Consortium. Crowdfunded whole-genome sequencing of the celebrity cat Lil BUB identifies causal mutations for her osteopetrosis and polydactyly. bioRxiv 2019, 556761. [Google Scholar] [CrossRef]
- Mauler, D.A.; Gandolfi, B.; Reinero, C.R.; Spooner, J.L.; Lyons, L.A.; 99 Lives Consortium. Precision Medicine in Cats: Novel Niemann-Pick Type C1 Diagnosed by Whole-Genome Sequencing. J. Vet. Intern. Med. 2017, 539–544. [Google Scholar] [CrossRef]
- den Dunnen, J.T.; Dalgleish, R.; Maglott, D.R.; Hart, R.K.; Greenblatt, M.S.; McGowan-Jordan, J.; Roux, A.-F.; Smith, T.; Antonarakis, S.E.; Taschner, P.E.M. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum. Mutat. 2016, 37, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLOS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Sheth, N.; Roca, X.; Hastings, M.L.; Roeder, T.; Krainer, A.R.; Sachidanandam, R. Comprehensive splice-site analysis using comparative genomics. Nucleic Acids Res. 2006, 34, 3955–3967. [Google Scholar] [CrossRef]
- Nistala, H.; Mäkitie, O.; Jüppner, H. Caffey disease: New perspectives on old questions. Bone 2014, 60, 246–251. [Google Scholar] [CrossRef]
- Campbell, B.G.; Wootton, J.A.M.; MacLeod, J.N.; Minor, R.R. Sequence of Normal Canine COL1A1 cDNA 1 and Identification of a Heterozygous alpha1 (I) Collagen Gly208Ala Mutation in a Severe Case of Canine Osteogenesis Imperfecta. Arch. Biochem. Biophys. 2000, 384, 37–46. [Google Scholar] [CrossRef]
- Ha, B.G.; Hong, J.M.; Park, J.-Y.; Ha, M.-H.; Kim, T.-H.; Cho, J.-Y.; Ryoo, H.-M.; Choi, J.-Y.; Shin, H.-I.; Chun, S.Y.; et al. Proteomic profile of osteoclast membrane proteins: Identification of Na+/H+ exchanger domain containing 2 and its role in osteoclast fusion. Proteomics 2008, 8, 2625–2639. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Hiraoka, S.; Furuichi, T.; Nishimura, G.; Shibata, S.; Yanagishita, M.; Rimoin, D.L.; Superti-Furga, A.; Nikkels, P.G.; Ogawa, M.; Katsuyama, K.; et al. Nucleotide-sugar transporter SLC35D1 is critical to chondroitin sulfate synthesis in cartilage and skeletal development in mouse and human. Nat. Med. 2007, 13, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, K.; Kitagawa, H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol. 2000, 10, 518–527. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affected Dog | Total Variants in the Whole Genome | Private Variants after Filtering against 584 Publically Available Genomes | ||||
|---|---|---|---|---|---|---|
| Total | Protein-Changing | Synonymous | Intronic | Intergenic | ||
| American Staffordshire Terrier (CHS) | 4,861,208 | 4582 | 76 (67/9) | 71 (69/2) | 2217 (1832/385) | 2218 (1831/387) |
| Australian Terrier (CMO) | 5,495,830 | 13,147 | 79 (73/6) | 30 (24/6) | 6527 (6012/515) | 6511 (5875/636) |
| Basset Hound (CMO) | 5,573,559 | 13,370 | 96 (87/9) | 30 (28/2) | 6535 (5917/618) | 6709 (6073/636) |
| Cairn Terrier (CMO) | 5,425,950 | 7373 | 55 (45/10) | 17 (16/1) | 3486 (3066/420) | 3815 (3344/471) |
| Curly Coated Retriever (CMO) | 5,326,349 | 4960 | 34 (34/0) | 15 (15/0) | 2405 (2248/157) | 2506 (2260/246) |
| German Wirehaired Pointer (CMO) | 5,519,945 | 11,713 | 63 (52/11) | 28 (25/3) | 5706 (5137/569) | 5916 (5291/625) |
| Old English Sheepdog (CMO) | 5,283,629 | 10,711 | 59 (52/7) | 22 (17/5) | 4966 (4175/791) | 5664 (4558/1106) |
| Weimaraner (CMO) | 5,201,857 | 8056 | 54 (48/6) | 26 (21/5) | 3859 (3183/676) | 4117 (3307/810) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Letko, A.; Leuthard, F.; Jagannathan, V.; Corlazzoli, D.; Matiasek, K.; Schweizer, D.; Hytönen, M.K.; Lohi, H.; Leeb, T.; Drögemüller, C. Whole Genome Sequencing Indicates Heterogeneity of Hyperostotic Disorders in Dogs. Genes 2020, 11, 163. https://doi.org/10.3390/genes11020163
Letko A, Leuthard F, Jagannathan V, Corlazzoli D, Matiasek K, Schweizer D, Hytönen MK, Lohi H, Leeb T, Drögemüller C. Whole Genome Sequencing Indicates Heterogeneity of Hyperostotic Disorders in Dogs. Genes. 2020; 11(2):163. https://doi.org/10.3390/genes11020163
Chicago/Turabian StyleLetko, Anna, Fabienne Leuthard, Vidhya Jagannathan, Daniele Corlazzoli, Kaspar Matiasek, Daniela Schweizer, Marjo K. Hytönen, Hannes Lohi, Tosso Leeb, and Cord Drögemüller. 2020. "Whole Genome Sequencing Indicates Heterogeneity of Hyperostotic Disorders in Dogs" Genes 11, no. 2: 163. https://doi.org/10.3390/genes11020163
APA StyleLetko, A., Leuthard, F., Jagannathan, V., Corlazzoli, D., Matiasek, K., Schweizer, D., Hytönen, M. K., Lohi, H., Leeb, T., & Drögemüller, C. (2020). Whole Genome Sequencing Indicates Heterogeneity of Hyperostotic Disorders in Dogs. Genes, 11(2), 163. https://doi.org/10.3390/genes11020163