Cascade Testing for Fragile X Syndrome in a Rural Setting in Cameroon (Sub-Saharan Africa)

 ,
,
Abstract
1. Introduction
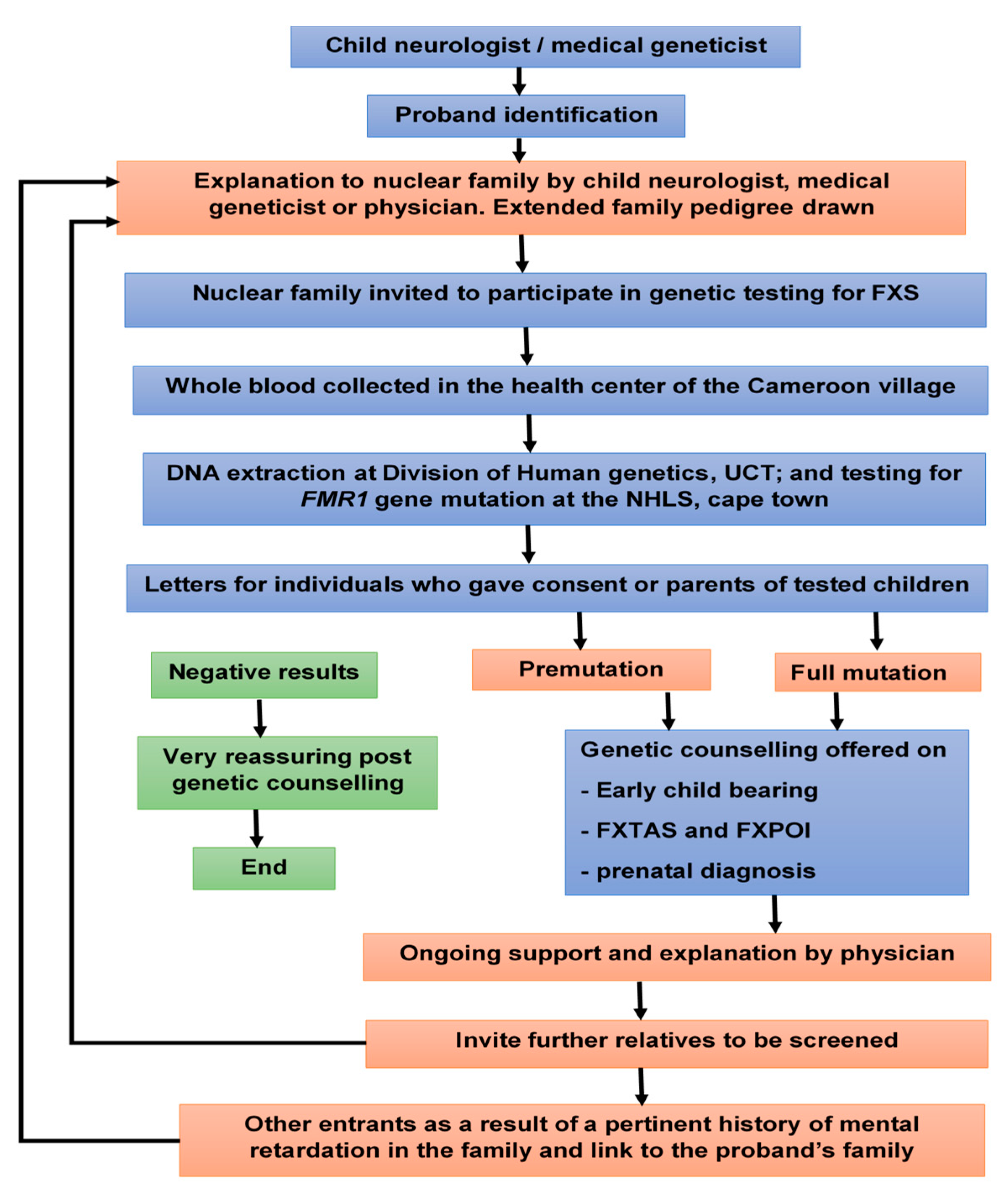
2. Material and Methods
2.1. Ethical Approvals
2.2. Participants
2.3. Molecular Analysis
2.4. Statistical Analysis
3. Results
3.1. Participants Sociodemographic
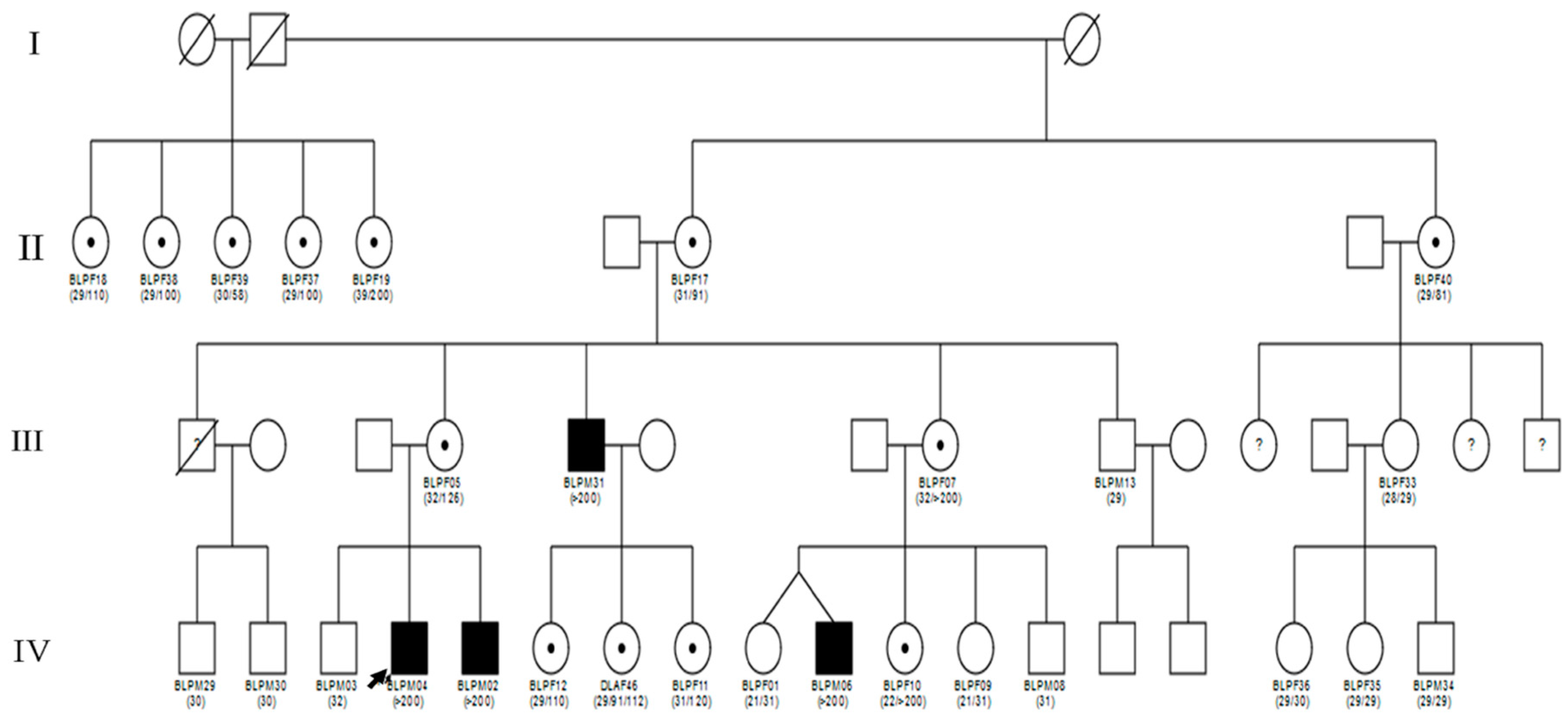
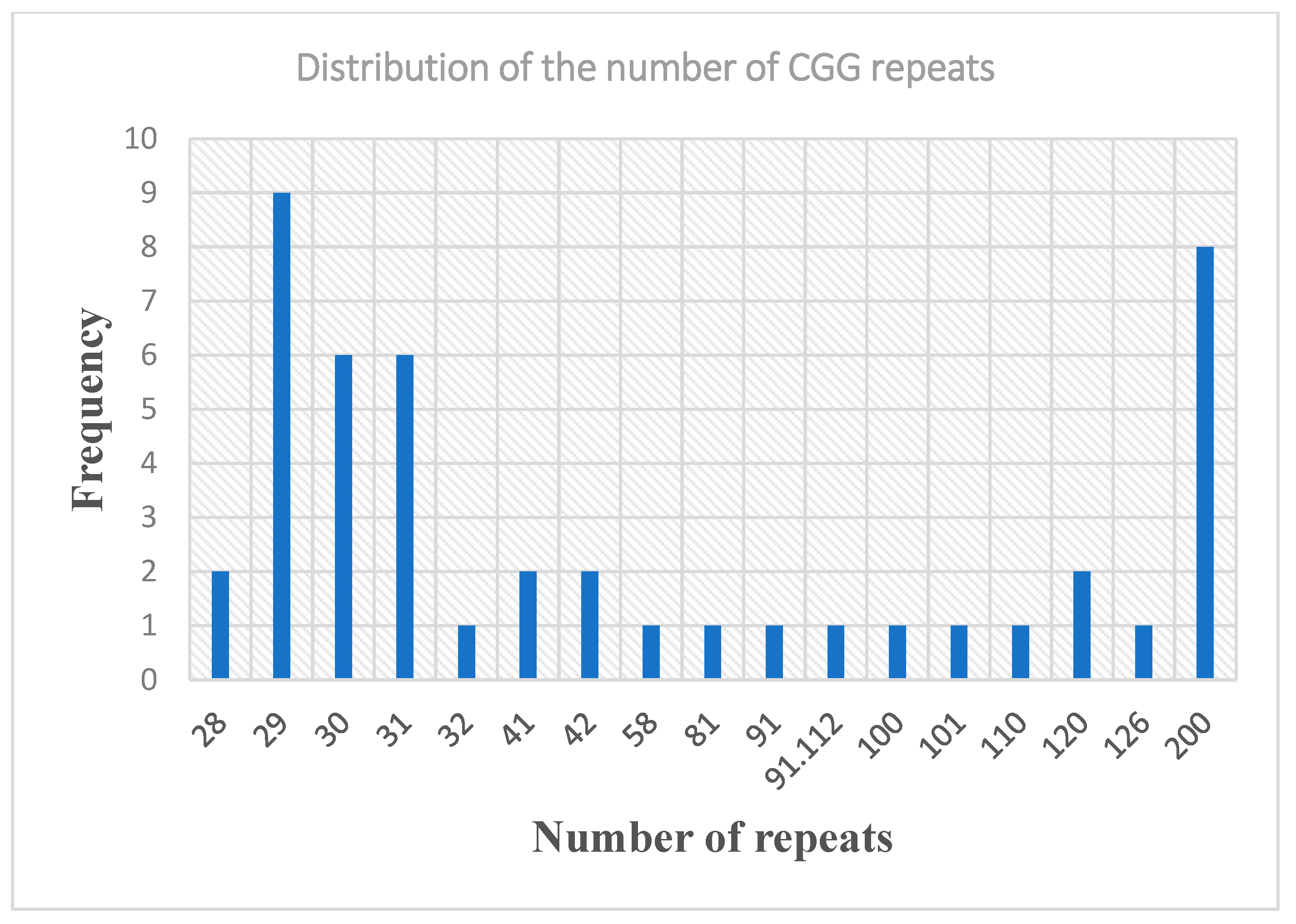
3.2. Molecular and Pedigree Analysis
4. Discussion
4.1. Practice Implications
4.2. Research Recommendation
4.3. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. Part A 2014, 164, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Krause, S.E.; Wuu, J.; Leurgans, S.; Guter, S.J.; Block, S.S.; Salt, J.; Cook, E.; Maino, D.M.; Berry-Kravis, E. Vocabulary comprehension in adults with fragile X syndrome (FXS). J. Neurodev. Disord. 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.L.; Freund, L. Behavioral phenotype of fragile X syndrome: DSM-III-R autistic behavior in male children. Am. J. Med. Genet. 1992, 43, 35–46. [Google Scholar] [CrossRef]
- Thurman, A.J.; McDuffie, A.; Hagerman, R.J.; Josol, C.K.; Abbeduto, L. Language Skills of Males with Fragile X Syndrome or Nonsyndromic Autism Spectrum Disorder. J. Autism Dev. Disord. 2017, 47, 728–743. [Google Scholar] [CrossRef]
- Gabis, L.V.; Hochberg, O.; Attia, O.L.; Banet-Levi, Y.; Topf, D.; Shefer, S. Prolonged Time Lag to Final Diagnosis of Fragile X Syndrome. J. Pediatr. 2018, 193, 217–221. [Google Scholar] [CrossRef]
- Lachiewicz, A.M.; Dawson, D.V.; Spiridigliozzi, G.A. Physical characteristics of young boys with fragile X syndrome: Reasons for difficulties in making a diagnosis in young males. Am. J. Med. Genet. 2000, 92, 229–236. [Google Scholar] [CrossRef]
- Jin, P.; Warren, S.T. Understanding the molecular basis of fragile X syndrome. Hum. Mol. Genet. 2000, 9, 901–908. [Google Scholar] [CrossRef]
- Santoro, M.R.; Bray, S.M.; Warren, S.T. Molecular Mechanisms of Fragile X Syndrome: A Twenty-Year Perspective. Annu. Rev. Pathol. Mech. Dis. 2012, 7, 219–245. [Google Scholar] [CrossRef]
- Crawford, D.C.; Acu, J.M.; Sherman, S.L. FMR1 and the fragile X syndrome: Human genome epidemiology review. Genet. Med. 2001, 3, 359–371. [Google Scholar] [CrossRef]
- Lozano, R.; Rosero, C.A.; Hagerman, R.J. Fragile X spectrum disorders. Intractable Rare Dis. Res. 2014, 3, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.; Han, Y.; Dy, A.B.C.; Du, J.; Jin, H.; Qin, J.; Zhang, J.; Li, Q.; Hagerman, R.J. Fragile X Syndrome: Prevalence, Treatment, and Prevention in China. Front. Neurol. 2017, 8, 254. [Google Scholar] [CrossRef] [PubMed]
- Maurin, T.; Zongaro, S.; Bardoni, B. Fragile X Syndrome: From molecular pathology to therapy. Neurosci. Biobehav. Rev. 2014, 46, 242–255. [Google Scholar] [CrossRef]
- Qurashi, A.; Chang, S.; Jin, P. Role of microRNA Pathway in Mental Retardation. Sci. World J. 2007, 7, 146–154. [Google Scholar] [CrossRef]
- Peprah, E. Fragile X syndrome: The FMR1 CGG repeat distribution among world populations. Ann. Hum. Genet. 2012, 76, 178–191. [Google Scholar] [CrossRef]
- Essop, F.B.; Krause, A. Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-1-related disorders in Johannesburg, South Africa: A 20-year review. S. Afr. Med. J. 2013, 103, 994. [Google Scholar] [CrossRef]
- Meguid, N.A.; Ismail, M.F.; El-Mahdy, R.S.; Barakat, M.A.; El-Awady, M.K. Simple molecular diagnostic method for fragile X syndrome in Egyptian patients: Pilot study. Acta Biochim. Pol. 2014, 61, 259–263. [Google Scholar] [CrossRef]
- Wonkam, A.; Tekendo, C.N.; Sama, D.J.; Zambo, H.; Dahoun, S.; Béna, F.; Morris, M.A. Initiation of a medical genetics service in sub-Saharan Africa: Experience of prenatal diagnosis in Cameroon. Eur. J. Med. Genet. 2011, 54, e399–e404. [Google Scholar] [CrossRef]
- Kromberg, J.G.; Sizer, E.B.; Christianson, A.L. Genetic services and testing in South Africa. J. Community Genet. 2013, 4, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Lubala, T.K.; Lumaka, A.; Mbuyi-Musanzayi, S.; Kayembe, T.; Shongo, M.Y.; Mukuku, O.; Lubala, N.; Malamba-Lez, D.; Luboya, O.N.; Lukusa-Tshilobo, P. Fragile X syndrome with mosaic size mutation in a Bantu patient from Central Africa. Clin. Dysmorphol. 2018, 27, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Lumaka, A.; Lubala, T.K.; Race, V.; Peeters, H.; Lukusa, P.; Devriendt, K. Usefulness of fragile X checklist and CGG distribution in specialized institutions in Kinshasa, DR Congo. J. Community Genet. 2019, 10, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Nguefack, S.; Kamga, K.K.; Moifo, B.; Chiabi, A.; Mah, E.; Mbonda, E. Causes of developmental delay in children of 5 to 72 months old at the child neurology unit of Yaounde Gynaeco-Obstetric and Paediatric Hospital (Cameroon). Open J. Pediatr. 2013, 3, 279. [Google Scholar] [CrossRef]
- Population, Total—Cameroon|Data 2018. Available online: https://data.worldbank.org/indicator/SP.POP.TOTL?locations=CM (accessed on 10 January 2020).
- Tishkoff, S.A.; Reed, F.A.; Friedlaender, F.R.; Ehret, C.; Ranciaro, A.; Froment, A.; Hirbo, J.B.; Awomoyi, A.A.; Bodo, J.-M.; Doumbo, O.; et al. The genetic structure and history of Africans and African Americans. Science 2009, 324, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- World-Bank. Education for All-Fast Track Initiative: Support to the Education Sector; Report No 48373; World-Bank: Washington, DC, USA, 2010; pp. 1–2. [Google Scholar]
- Wonkam, A.; Bitoungui, V.J.N.; Vorster, A.A.; Ramesar, R.; Cooper, R.S.; Tayo, B.; Lettre, G.; Ngogang, J. Association of Variants at BCL11A and HBS1L-MYB with Hemoglobin F and Hospitalization Rates among Sickle Cell Patients in Cameroon. PLoS ONE 2014, 9, e92506. [Google Scholar] [CrossRef]
- Wonkam, A.; Njamnshi, A.K.; Angwafo, F.F.; Iii, F.F.A. Knowledge and attitudes concerning medical genetics amongst physicians and medical students in Cameroon (sub-Saharan Africa). Genet. Med. 2006, 8, 331. [Google Scholar] [CrossRef]
- Wonkam, A.; De Vries, J. Returning incidental findings in African genomics research. Nat. Genet. 2019, 52, 1–4. [Google Scholar] [CrossRef]
- NIH. NCI Dictionary of Genetics Terms: Cascade Testing 2019. Available online: https://www.cancer.gov/publications/dictionaries/genetics-dictionary/def/cascade-screening (accessed on 17 December 2019).
- Hampel, H. Genetic counseling and cascade genetic testing in Lynch syndrome. Fam. Cancer 2016, 15, 423–427. [Google Scholar] [CrossRef]
- McClaren, B.J.; Aitken, M.; Massie, J.; Amor, D.; Ukoumunne, O.C.; Metcalfe, S.A. Cascade carrier testing after a child is diagnosed with cystic fibrosis through newborn screening: Investigating why most relatives do not have testing. Genet. Med. 2013, 15, 533. [Google Scholar] [CrossRef]
- Newson, A.J.; Humphries, S.E.; Newson, S.E.H.A.J. Cascade testing in familial hypercholesterolaemia: How should family members be contacted? Eur. J. Hum. Genet. 2005, 13, 401. [Google Scholar] [CrossRef]
- Boat, T.F.; Wu, J.T. Clinical Characteristics of Intellectual Disabilities; National Academies Press: Washington, DC, USA, 2015. Available online: https://www.ncbi.nlm.nih.gov/books/NBK332877/ (accessed on 20 June 2019).
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®), 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013; 970p. [Google Scholar]
- Nolin, S.L.; Brown, W.T.; Glicksman, A.; Houck, J.G.E.; Gargano, A.D.; Sullivan, A.; Biancalana, V.; Bröndum-Nielsen, K.; Hjalgrim, H.; Holinski-Feder, E.; et al. Expansion of the Fragile X CGG Repeat in Females with Premutation or Intermediate Alleles. Am. J. Hum. Genet. 2003, 72, 454–464. [Google Scholar] [CrossRef]
- Biancalana, V.; Glaeser, D.; McQuaid, S.; Steinbach, P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet. 2015, 23, 417. [Google Scholar] [CrossRef] [PubMed]
- Renda, M.M.; Voigt, R.G.; Babovic-Vuksanovic, D.; Highsmith, W.E.; Vinson, S.S.; Sadowski, C.M.; Hagerman, R.J. Neurodevelopmental disabilities in children with intermediate and premutation range fragile X cytosine-guanine-guanine expansions. J. Child Neurol. 2014, 29, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-C.; Lee, C.-N.; Wang, Y.-C.; Chen, C.-L.; Lin, T.-K.; Su, Y.-N.; Lin, M.-W.; Kang, J.; Tai, Y.-Y.; Hsu, W.-W.; et al. Fragile X syndrome carrier screening in pregnant women in Chinese Han population. Sci. Rep. 2019, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.S.; Leung, K. Challenges in prenatal screening and counselling for fragile X syndrome. Hong Kong Med. J. 2017, 23, 108–109. [Google Scholar] [CrossRef] [PubMed]
- Ciaccio, C.; Fontana, L.; Milani, D.; Tabano, S.; Miozzo, M.; Esposito, S. Fragile X syndrome: A review of clinical and molecular diagnoses. Ital. J. Pediatr. 2017, 43, 39. [Google Scholar] [CrossRef] [PubMed]
- Heine-Suñer, D.; Torres-Juan, L.; Morlà, M.; Busquets, X.; Barcelo, F.; Picó, G.; Bonilla, L.; Govea, N.; Bernués, M.; Rosell, J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: A female member with complete inactivation of the functional X chromosome. Am. J. Med. Genet. Part A 2003, 122, 108–114. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the Fragile X–Associated Tremor/Ataxia Syndrome in a Premutation Carrier Population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef]
- Sherman, S.; Pletcher, B.A.; Driscoll, D.A. Fragile X syndrome: Diagnostic and carrier testing. Genet. Med. 2005, 7, 584–587. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variable | Mutation Pattern | Total | |||
|---|---|---|---|---|---|
| N | PM | FM | |||
| Subject | Male | 15 | 0 | 4 | 19 |
| Female | 13 | 10 | 4 | 27 | |
| ID Status of Males | Absent | 14 | 0 | 0 | 14 |
| Mild ID | 1 | 0 | 0 | 1 | |
| Severe ID | 0 | 0 | 4 | 4 | |
| ID Status of Females | Absent | 13 | 8 | 1 | 22 |
| Mild ID | 0 | 2 | 3 | 5 | |
| Severe ID | 0 | 0 | 0 | 0 | |
| Maternal Allele (CGG Repeat) Transmitted | Child Allele (CGG rRpeat) | CGG Repeat Increase | Maternal Allele (CGG Repeats) Not Transmitted | |
|---|---|---|---|---|
| Male | Female | |||
| 91 (I) | 126 | +35 | 31 | |
| >200 | +>109 | 31 | ||
| >200 | +>109 | 31 | ||
| 126 (II) | >200 | +>74 | 32 | |
| >200 | +>74 | 32 | ||
| >200 (III) | >200 | 0 | 32 | |
| >200 | 0 | 32 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kengne Kamga, K.; Nguefack, S.; Minka, K.; Wonkam Tingang, E.; Esterhuizen, A.; Nchangwi Munung, S.; De Vries, J.; Wonkam, A. Cascade Testing for Fragile X Syndrome in a Rural Setting in Cameroon (Sub-Saharan Africa). Genes 2020, 11, 136. https://doi.org/10.3390/genes11020136
Kengne Kamga K, Nguefack S, Minka K, Wonkam Tingang E, Esterhuizen A, Nchangwi Munung S, De Vries J, Wonkam A. Cascade Testing for Fragile X Syndrome in a Rural Setting in Cameroon (Sub-Saharan Africa). Genes. 2020; 11(2):136. https://doi.org/10.3390/genes11020136
Chicago/Turabian StyleKengne Kamga, Karen, Séraphin Nguefack, Khuthala Minka, Edmond Wonkam Tingang, Alina Esterhuizen, Syntia Nchangwi Munung, Jantina De Vries, and Ambroise Wonkam. 2020. "Cascade Testing for Fragile X Syndrome in a Rural Setting in Cameroon (Sub-Saharan Africa)" Genes 11, no. 2: 136. https://doi.org/10.3390/genes11020136
APA StyleKengne Kamga, K., Nguefack, S., Minka, K., Wonkam Tingang, E., Esterhuizen, A., Nchangwi Munung, S., De Vries, J., & Wonkam, A. (2020). Cascade Testing for Fragile X Syndrome in a Rural Setting in Cameroon (Sub-Saharan Africa). Genes, 11(2), 136. https://doi.org/10.3390/genes11020136