ALT: A Multi-Faceted Phenomenon


Abstract
1. Telomere Maintenance Mechanisms in Cancer
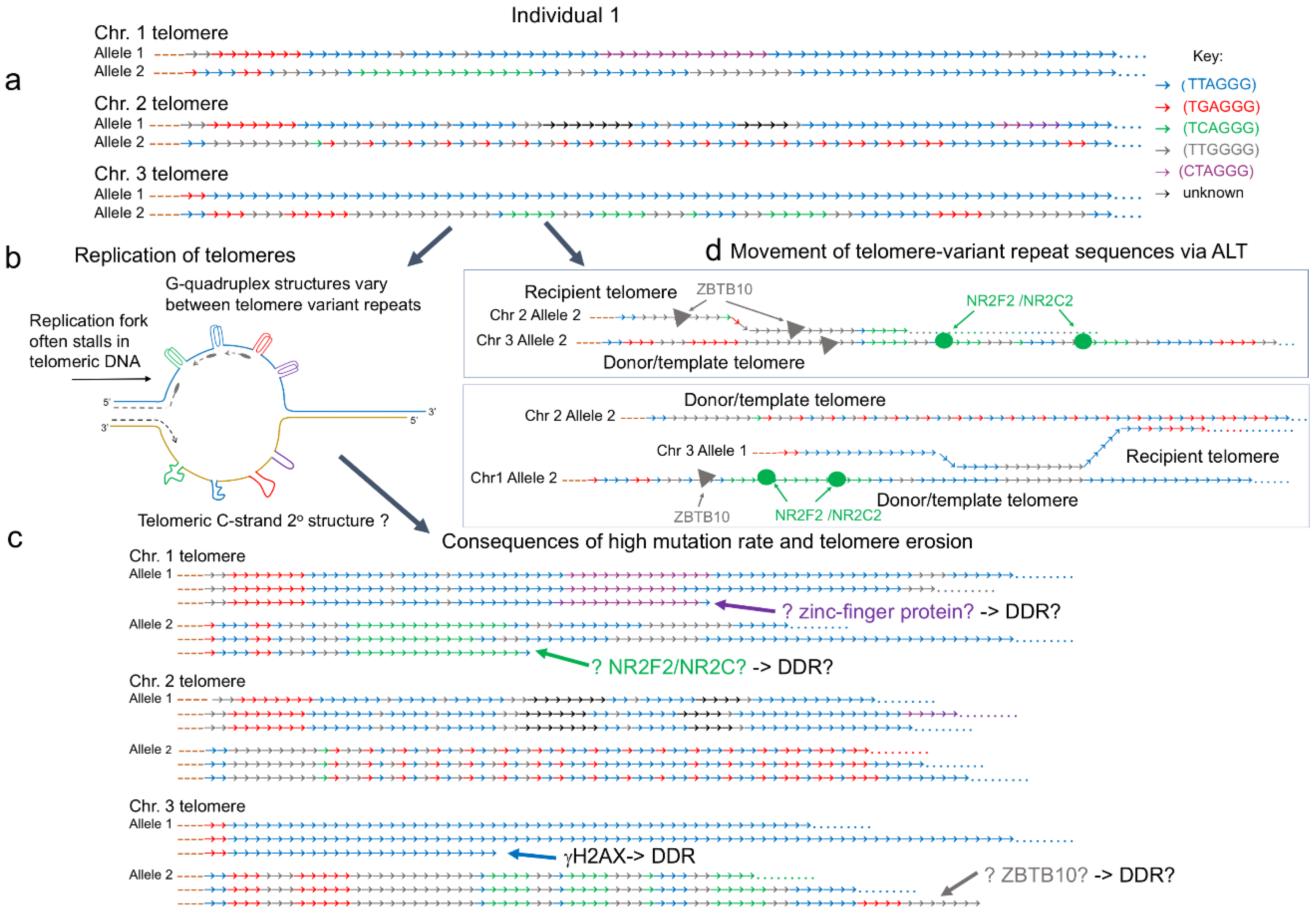
2. Phenotypes Specific to ALT
3. Degenerate Variant Repeats: A Role in ALT?
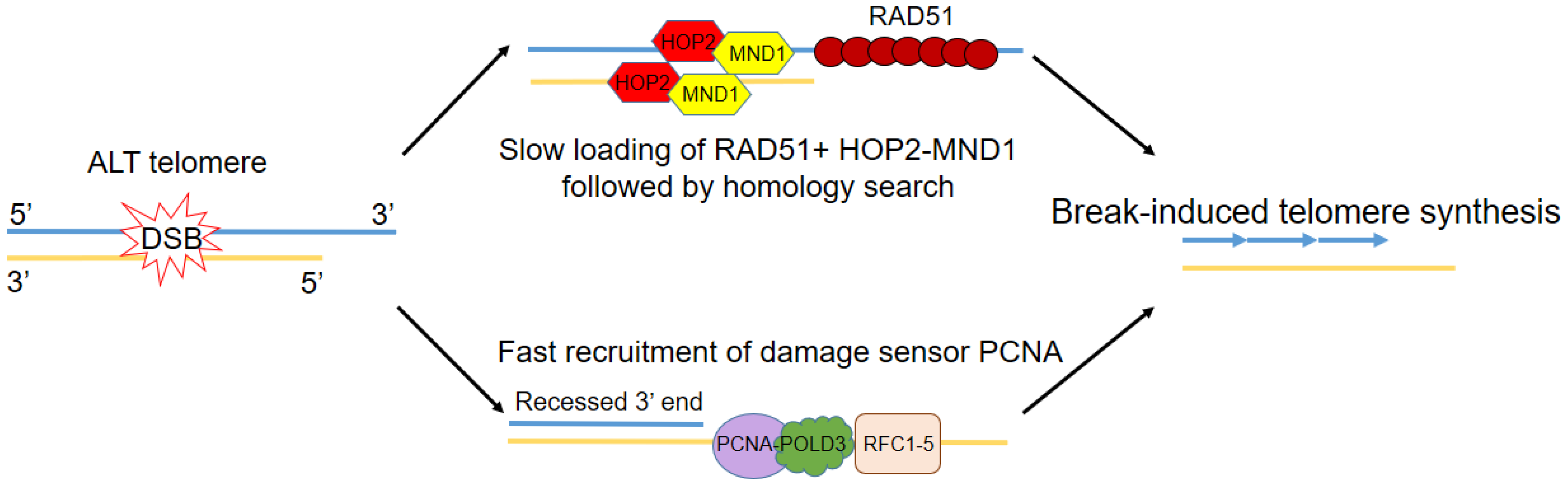
4. Pathways to Telomere Elongation in ALT and Relationship to Genome Instability
5. Therapeutic Outlook and Challenges
6. Concluding Remarks
Funding
Conflicts of Interest
References
- de Lange, T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Greenberg, R.A. ALTernative telomere maintenance and cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef]
- Reddel, R.R. Telomere maintenance mechanisms in cancer: Clinical implications. Curr. Pharm. Des. 2014, 20, 6361–6374. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. The telomere terminal transferase of Tetrahymena is a ribonucleoprotein enzyme with two kinds of primer specificity. Cell 1987, 51, 887–898. [Google Scholar] [CrossRef]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Bryan, T.M.; Englezou, A.; Dalla-Pozza, L.; Dunham, M.A.; Reddel, R.R. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997, 3, 1271–1274. [Google Scholar] [CrossRef]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive proliferation of human cancer cells with ever-shorter telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar]
- Liau, J.Y.; Lee, J.C.; Tsai, J.H.; Yang, C.Y.; Liu, T.L.; Ke, Z.L.; Hsu, H.H.; Jeng, Y.M. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Jeng, Y.M.; Liau, J.Y.; Tsai, J.H.; Hsu, H.H.; Yang, C.Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Henson, J.D.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Alternative lengthening of telomeres in mammalian cells. Oncogene 2002, 21, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Bechter, O.E.; Zou, Y.; Walker, W.; Wright, W.E.; Shay, J.W. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004, 64, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Hwang, S.S.; Liesa, M.; Gan, B.; Sahin, E.; Jaskelioff, M.; Ding, Z.; Ying, H.; Boutin, A.T.; Zhang, H.; et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 2012, 148, 651–663. [Google Scholar] [CrossRef]
- Queisser, A.; Heeg, S.; Thaler, M.; von Werder, A.; Opitz, O.G. Inhibition of telomerase induces alternative lengthening of telomeres during human esophageal carcinogenesis. Cancer Genet. 2013, 206, 374–386. [Google Scholar] [CrossRef]
- Lafferty-Whyte, K.; Cairney, C.J.; Will, M.B.; Serakinci, N.; Daidone, M.G.; Zaffaroni, N.; Bilsland, A.; Keith, W.N. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009, 28, 3765–3774. [Google Scholar] [CrossRef]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef]
- Coluzzi, E.; Buonsante, R.; Leone, S.; Asmar, A.J.; Miller, K.L.; Cimini, D.; Sgura, A. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci. Rep. 2017, 7, 43309. [Google Scholar] [CrossRef] [PubMed]
- De Vitis, M.; Berardinelli, F.; Coluzzi, E.; Marinaccio, J.; O’Sullivan, R.J.; Sgura, A. X-rays activate telomeric homologous recombination mediated repair in primary cells. Cells 2019, 8, 708. [Google Scholar] [CrossRef] [PubMed]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2015, 6, 7493–7503. [Google Scholar] [CrossRef] [PubMed]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Griffith, J.D. Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol. Cell. Biol. 2004, 24, 9948–9957. [Google Scholar] [CrossRef]
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Reddel, R.R. The role of telomere trimming in normal telomere length dynamics. Cell Cycle 2012, 11, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Oganesian, L.; Karlseder, J. Mammalian 5′ C-rich telomeric overhangs are a mark of recombination-dependent telomere maintenance. Mol. Cell 2011, 42, 224–236. [Google Scholar] [CrossRef]
- Oganesian, L.; Karlseder, J. 5′ C-rich telomeric overhangs are an outcome of rapid telomere truncation events. DNA Repair (Amst) 2013, 12, 238–245. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, Z.; Shengzhao, G.; Li, X.; Liu, H.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925. [Google Scholar] [CrossRef]
- Henson, J.D.; Cao, Y.; Huschtscha, L.I.; Chang, A.C.; Au, A.Y.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185. [Google Scholar] [CrossRef]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef]
- Chen, Y.A.; Shen, Y.L.; Hsia, H.Y.; Tiang, Y.P.; Sung, T.L.; Chen, L.Y. Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 2017, 24, 1124–1131. [Google Scholar] [CrossRef]
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londoño-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.Q.; Zhong, Z.H.; Henson, J.D.; Neumann, A.A.; Chang, A.C.; Reddel, R.R. Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of the MRE11/RAD50/NBS1 complex. Mol. Cell Biol. 2005, 25, 2708–2721. [Google Scholar] [CrossRef] [PubMed]
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179. [Google Scholar]
- Jamiruddin, M.R.; Kaitsuka, T.; Hakim, F.; Fujimura, A.; Wei, F.Y.; Saitoh, H.; Tomizawa, K. HDAC9 regulates the alternative lengthening of telomere (ALT) pathway via the formation of ALT-associated PML bodies. Biochem. Biophys. Res. Commun. 2016, 481, 25–30. [Google Scholar] [CrossRef]
- Londoño-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327. [Google Scholar] [CrossRef] [PubMed]
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res. 2017, 45, 2615–2628. [Google Scholar] [CrossRef] [PubMed]
- Nabetani, A.; Ishikawa, F. Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol. Cell Biol. 2009, 29, 703–713. [Google Scholar] [CrossRef]
- Jeyapalan, J.N.; Varley, H.; Foxon, J.L.; Pollock, R.E.; Jeffreys, A.J.; Henson, J.D.; Reddel, R.R.; Royle, N.J. Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum. Mol. Genet. 2005, 14, 1785–1794. [Google Scholar] [CrossRef][Green Version]
- Jeyapalan, J.N.; Mendez-Bermudez, A.; Zaffaroni, N.; Dubrova, Y.E.; Royle, N.J. Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int. J. Cancer 2008, 122, 2414–2421. [Google Scholar] [CrossRef] [PubMed]
- Allshire, R.C.; Dempster, M.; Hastie, N.D. Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res. 1989, 17, 4611–4627. [Google Scholar] [CrossRef]
- Baird, D.M.; Jeffreys, A.J.; Royle, N.J. Mechanisms underlying telomere repeat turnover, revealed by hypervariable variant repeat distribution patterns in the human Xp/Yp telomere. EMBO J. 1995, 14, 5433–5443. [Google Scholar] [CrossRef] [PubMed]
- Pickett, H.A.; Baird, D.M.; Hoff-Olsen, P.; Meling, G.I.; Rognum, T.O.; Shaw, J.; West, K.P.; Royle, N.J. Telomere instability detected in sporadic colon cancers, some showing mutations in a mismatch repair gene. Oncogene 2004, 23, 3434–3443. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mendez-Bermudez, A.; Hills, M.; Pickett, H.A.; Phan, A.T.; Mergny, J.-L.; Riou, J.-F.; Royle, N.J. Human telomeres that contain (CTAGGG)n repeats show replication dependent instability in somatic cells and the male germline. Nucleic Acids Res. 2009, 37, 6225–6238. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hills, M.; Conomos, D.; Stutz, M.D.; Dagg, R.A.; Lau, L.M.; Reddel, R.R.; Pickett, H.A. Telomere extension by telomerase and ALT generates variant repeats by mechanistically distinct processes. Nucleic Acids Res. 2014, 42, 1733–1746. [Google Scholar] [CrossRef]
- Varley, H.; Pickett, H.A.; Foxon, J.L.; Reddel, R.R.; Royle, N.J. Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat. Genet. 2002, 30, 301–305. [Google Scholar] [CrossRef]
- Lee, M.; Teber, E.T.; Holmes, O.; Nones, K.; Patch, A.M.; Dagg, R.A.; Lau, L.M.S.; Lee, J.H.; Napier, C.E.; Arthur, J.W.; et al. Telomere sequence content can be used to determine ALT activity in tumours. Nucleic Acids Res. 2018, 46, 4903–4918. [Google Scholar] [CrossRef]
- Déjardin, J.; Kingston, R.E. Purification of proteins associated with specific genomic Loci. Cell 2009, 136, 175–186. [Google Scholar] [CrossRef]
- Conomos, D.; Stutz, M.D.; Hills, M.; Neumann, A.A.; Bryan, T.M.; Reddel, R.R.; Pickett, H.A. Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J. Cell Biol. 2012, 199, 893–906. [Google Scholar] [CrossRef]
- Conomos, D.; Reddel, R.R.; Pickett, H.A. NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat. Struct. Mol. Biol. 2014, 21, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Alhendi, A.S.N.; Royle, N.J.R. The nuclear receptor NR2F2 regulates the cell cycle but only plays an indirect role in the Alternative Lengthening of Telomeres. (manuscript in preparation).
- Xu, M.; Qin, J.; Wang, L.; Lee, H.J.; Kao, C.Y.; Liu, D.; Songyang, Z.; Chen, J.; Tsai, M.J.; Tsai, S.Y. Nuclear receptors regulate alternative lengthening of telomeres through a novel noncanonical FANCD2 pathway. Sci. Adv. 2019, 5, eaax6366. [Google Scholar] [CrossRef] [PubMed]
- Marzec, P.; Armenise, C.; Pérot, G.; Roumelioti, F.M.; Basyuk, E.; Gagos, S.; Chibon, F.; Déjardin, J. Nuclear-receptor-mediated telomere insertion leads to genome instability in ALT cancers. Cell 2015, 160, 913–927. [Google Scholar] [CrossRef] [PubMed]
- Bluhm, A.; Viceconte, N.; Li, F.; Rane, G.; Ritz, S.; Wang, S.; Levin, M.; Shi, Y.; Kappei, D.; Butter, F. ZBTB10 binds the telomeric variant repeat TTGGGG and interacts with TRF2. Nucleic Acids Res. 2019, 47, 1896–1907. [Google Scholar] [CrossRef]
- Malkova, A.; Ivanov, E.L.; Haber, J.E. Double-strand break repair in the absence of RAD51 in yeast: A possible role for break-induced DNA replication. Proc. Natl. Acad. Sci. USA 1996, 93, 7131–7136. [Google Scholar] [CrossRef]
- McEachern, M.J.; Haber, J.E. Break-induced replication and recombinational telomere elongation in yeast. Annu. Rev. Biochem. 2006, 75, 111–135. [Google Scholar] [CrossRef]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar] [CrossRef]
- Pickett, H.A.; Reddel, R.R. Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat. Struct. Mol. Biol. 2015, 22, 875–880. [Google Scholar] [CrossRef]
- Lydeard, J.R.; Jain, S.; Yamaguchi, M.; Haber, J.E. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 2007, 448, 820–823. [Google Scholar] [CrossRef]
- Mendez-Bermudez, A.; Hidalgo-Bravo, A.; Cotton, V.E.; Gravani, A.; Jeyapalan, J.N.; Royle, N.J. The roles of WRN and BLM RecQ helicases in the Alternative Lengthening of Telomeres. Nucleic Acids Res. 2012, 40, 10809–10820. [Google Scholar] [CrossRef]
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Pentz, R.; Figueira, A.M.; Arora, R.; Lee, Y.W.; Hodson, C.; Wischnewski, H.; Deans, A.J.; Azzalin, C.M. FANCM limits ALT activity by restricting telomeric replication stress induced by deregulated BLM and R-loops. Nat. Commun. 2019, 10, 2253. [Google Scholar] [CrossRef] [PubMed]
- Cho, N.W.; Dilley, R.L.; Lampson, M.A.; Greenberg, R.A. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 2014, 159, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Dilley, R.L.; Verma, P.; Cho, N.W.; Winters, H.D.; Wondisford, A.R.; Greenberg, R.A. Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 2016, 539, 54–58. [Google Scholar] [CrossRef]
- Min, J.; Wright, W.E.; Shay, J.W. Alternative lengthening of telomeres mediated by mitotic DNA synthesis engages break-induced replication processes. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative lengthening of telomeres through two distinct break-induced replication pathways. Cell Rep. 2019, 26, 955–968.e953. [Google Scholar] [CrossRef]
- Sobinoff, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D.; Reddel, R.R.; Pickett, H.A. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; de Lange, T.; De, S.; Petrini, J.H.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 2012, 8, e1002772. [Google Scholar] [CrossRef]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538. [Google Scholar] [CrossRef]
- Episkopou, H.; Draskovic, I.; Van Beneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; Napier, C.E.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.E.; Maréchal, A.; Flynn, R.L. SMARCAL1 resolves replication stress at ALT telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and kataegis induced by telomere crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Ritchie, K.; Seah, C.; Moulin, J.; Isaac, C.; Dick, F.; Bérubé, N.G. Loss of ATRX leads to chromosome cohesion and congression defects. J. Cell Biol. 2008, 180, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Scheel, C.; Schaefer, K.L.; Jauch, A.; Keller, M.; Wai, D.; Brinkschmidt, C.; van Valen, F.; Boecker, W.; Dockhorn-Dworniczak, B.; Poremba, C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001, 20, 3835–3844. [Google Scholar] [CrossRef]
- Montgomery, E.; Argani, P.; Hicks, J.L.; DeMarzo, A.M.; Meeker, A.K. Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am. J. Pathol. 2004, 164, 1523–1529. [Google Scholar] [CrossRef]
- Costa, A.; Daidone, M.G.; Daprai, L.; Villa, R.; Cantù, S.; Pilotti, S.; Mariani, L.; Gronchi, A.; Henson, J.D.; Reddel, R.R.; et al. Telomere maintenance mechanisms in liposarcomas: Association with histologic subtypes and disease progression. Cancer Res. 2006, 66, 8918–8924. [Google Scholar] [CrossRef]
- Sakellariou, D.; Chiourea, M.; Raftopoulou, C.; Gagos, S. Alternative lengthening of telomeres: Recurrent cytogenetic aberrations and chromosome stability under extreme telomere dysfunction. Neoplasia 2013, 15, 1301–1313. [Google Scholar] [CrossRef]
- Dangoor, A.; Seddon, B.; Gerrand, C.; Grimer, R.; Whelan, J.; Judson, I. UK guidelines for the management of soft tissue sarcomas. Clin. Sarcoma Res. 2016, 6, 20. [Google Scholar] [CrossRef]
- Zhong, Z.H.; Jiang, W.Q.; Cesare, A.J.; Neumann, A.A.; Wadhwa, R.; Reddel, R.R. Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J. Biol. Chem. 2007, 282, 29314–29322. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]
- Salloum, R.; Hummel, T.R.; Kumar, S.S.; Dorris, K.; Li, S.; Lin, T.; Daryani, V.M.; Stewart, C.F.; Miles, L.; Poussaint, T.Y.; et al. A molecular biology and phase II study of imetelstat (GRN163L) in children with recurrent or refractory central nervous system malignancies: A pediatric brain tumor consortium study. J. Neurooncol. 2016, 129, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Chiappori, A.A.; Kolevska, T.; Spigel, D.R.; Hager, S.; Rarick, M.; Gadgeel, S.; Blais, N.; Von Pawel, J.; Hart, L.; Reck, M.; et al. A randomized phase II study of the telomerase inhibitor imetelstat as maintenance therapy for advanced non-small-cell lung cancer. Ann. Oncol. 2015, 26, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Episkopou, H.; Diman, A.; Claude, E.; Viceconte, N.; Decottignies, A. TSPYL5 depletion induces specific death of ALT cells through USP7-dependent proteasomal degradation of POT1. Mol. Cell 2019, 75, 469–482.e466. [Google Scholar] [CrossRef] [PubMed]
- Bakhos-Douaihy, D.; Desmaze, C.; Jeitany, M.; Gauthier, L.R.; Biard, D.; Junier, M.P.; Chneiweiss, H.; Boussin, F.D. ALT cancer cells are specifically sensitive to lysine acetyl transferase inhibition. Oncotarget 2019, 10, 773–784. [Google Scholar] [CrossRef]
- Zhang, X.W.; Yan, X.J.; Zhou, Z.R.; Yang, F.F.; Wu, Z.Y.; Sun, H.B.; Liang, W.X.; Song, A.X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef]
- Pompili, L.; Leonetti, C.; Biroccio, A.; Salvati, E. Diagnosis and treatment of ALT tumors: Is Trabectedin a new therapeutic option? J. Exp. Clin. Cancer Res. 2017, 36, 189. [Google Scholar] [CrossRef]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef]
- Huang, F.C.; Chang, C.C.; Wang, J.M.; Chang, T.C.; Lin, J.J. Induction of senescence in cancer cells by the G-quadruplex stabilizer, BMVC4, is independent of its telomerase inhibitory activity. Br. J. Pharmacol. 2012, 167, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Obata, S.; Yu, Z.; Bando, T.; Sugiyama, H. Recent progress of targeted g-quadruplex-preferred ligands toward cancer therapy. Molecules 2019, 24, 429. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.H.; Nie, X.; Fang, Y.; Zhang, Z.; Xiao, Y.; Mao, Z.; Liu, H.; Ren, J.; Wang, F.; Xia, L.; et al. A cisplatin derivative Tetra-Pt(bpy) as an oncotherapeutic agent for targeting ALT cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug Name | Reference | Mechanism of Action | Comments |
|---|---|---|---|
| Anacardic acid | [87] | Inhibits PCAF, a lysine acetyl transferase involved in regulation of APB-related genes | Natural product, not yet entered clinical trials |
| Arsenic Trioxide | [88] | Binds PML protein | Approved for use in the treatment of acute promyelocytic leukemia |
| CX-5461 | [93] | G-quadruplex stabilizer | Currently in clinical trial phase 1 |
| Mirin | [33,82,83] | Inhibits MRN | Small molecule inhibitor, has yet to progress to clinical trial stage |
| PIP-199 | [62] | Inhibitor of the MM2-RMI interaction within FANCM-BTR | Small molecule inhibitor, for research use only. Requires further studies before consideration for clinical trials |
| Quarfloxin | [93] | G-quadruplex stabilizer | Clinical trial terminated in phase 2 |
| Tetra-Pt(bpy) | [94] | Converts telomeric single-stranded DNA to G-quadruplex | Cisplatin derivative, not progressed to clinical trials yet |
| Trabectedin | [90] | Binds minor groove of DNA double helix | ALT positive cells found to be more sensitive than telomerase positive; reason unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sommer, A.; Royle, N.J. ALT: A Multi-Faceted Phenomenon. Genes 2020, 11, 133. https://doi.org/10.3390/genes11020133
Sommer A, Royle NJ. ALT: A Multi-Faceted Phenomenon. Genes. 2020; 11(2):133. https://doi.org/10.3390/genes11020133
Chicago/Turabian StyleSommer, Aurore, and Nicola J. Royle. 2020. "ALT: A Multi-Faceted Phenomenon" Genes 11, no. 2: 133. https://doi.org/10.3390/genes11020133
APA StyleSommer, A., & Royle, N. J. (2020). ALT: A Multi-Faceted Phenomenon. Genes, 11(2), 133. https://doi.org/10.3390/genes11020133