Resolving the Phylogeny of the Olive Family (Oleaceae): Confronting Information from Organellar and Nuclear Genomes

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Sequencing
2.2. Assembly of Cytoplasmic and Nuclear DNA Regions
2.2.1. Assembly of Plastome and Nuclear Ribosomal DNA (nrDNA) Cluster
2.2.2. Assembly of Mitochondrial Genes
2.2.3. Assembly of Genes Encoding Phytochromes
2.3. Phylogenetic Analyses
2.3.1. Phylogeny of Oleaceae Using Organellar DNA
2.3.2. Phylogeny of Oleaceae Using nrDNA
2.3.3. Phylogenetic Analyses of the Nuclear phy Gene Family
2.3.4. Phylogenetic Inference of Family Tree Using Data from Mixed Origin
3. Results
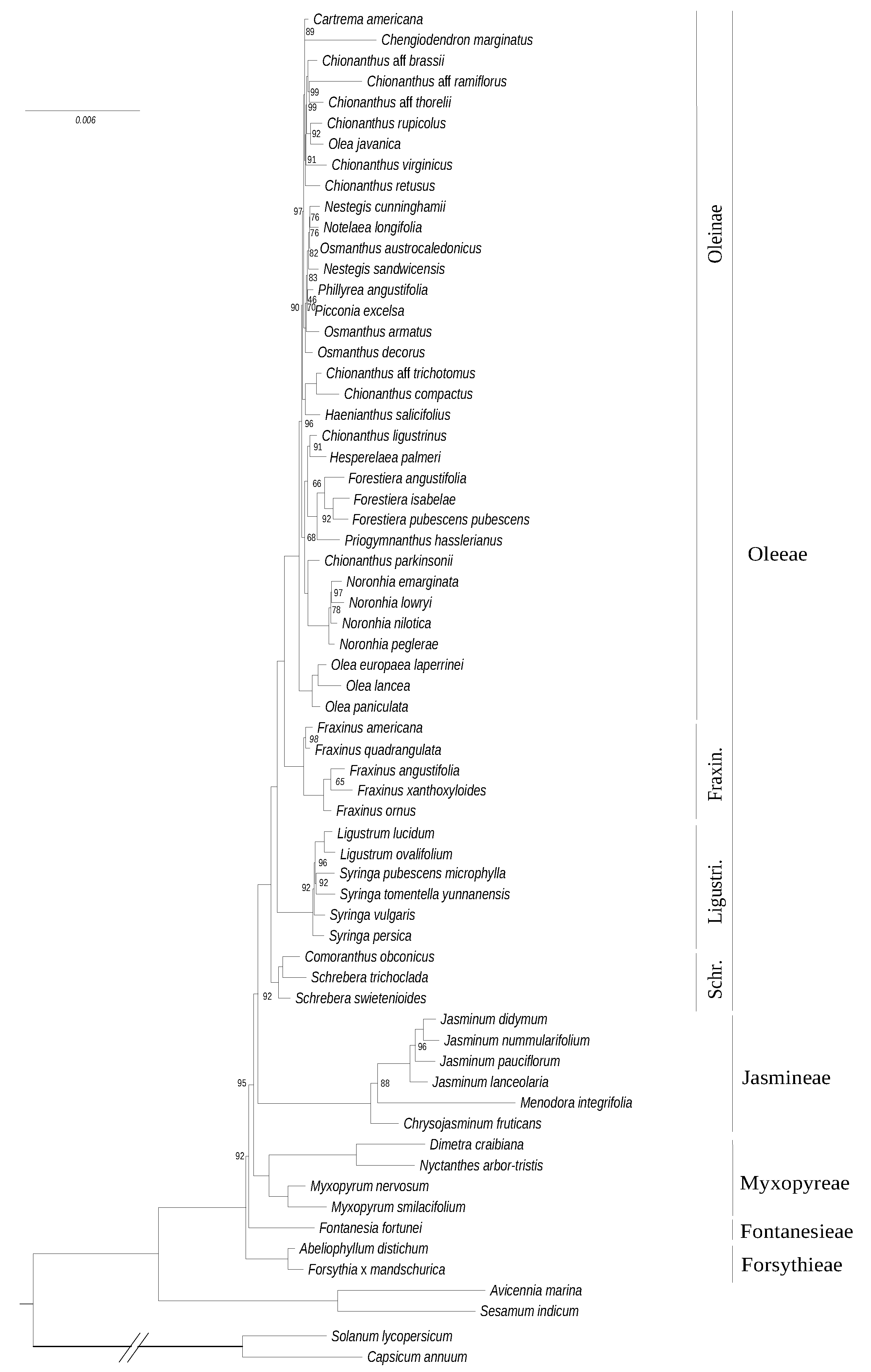
3.1. Phylogenetic Reconstructions Based on Chloroplast and Mitochondrial Genes
3.2. Phylogeny Based on the Nuclear Ribosomal Cluster
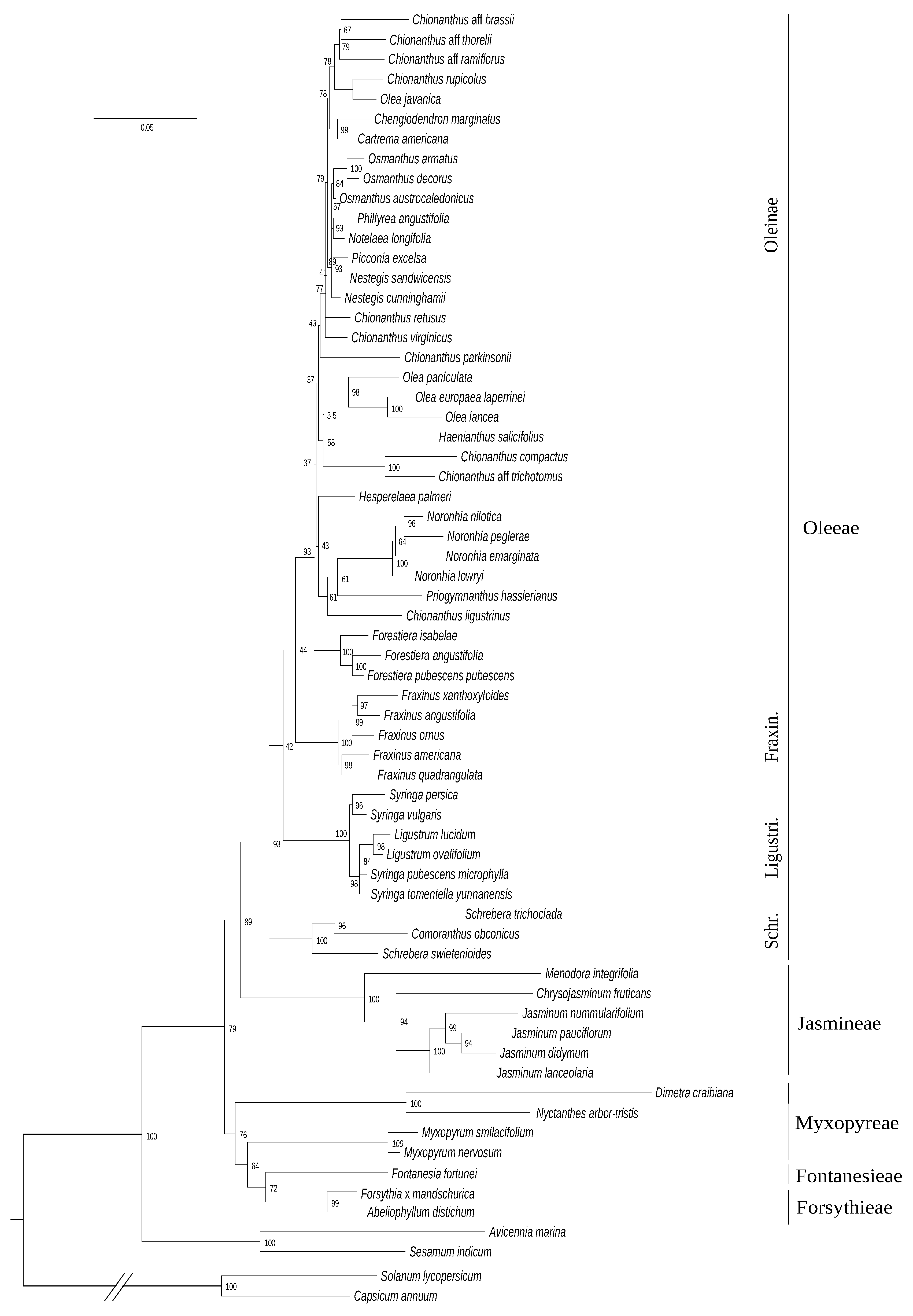
3.3. Phylogeny Based on Nuclear phy Gene Family
3.4. Phylogenetic Reconstruction Combining the Four Genomic Datasets
4. Discussion
4.1. Taxonomy of Oleaceae
4.2. Nuclear Gene Orthology and Polyploidization Events in Oleaceae
5. Concluding Remarks and Future Directions in Oleaceae Phylogenomics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Green, P.S. Oleaceae. In The Families and Genera of Vascular Plants, Flowering Plants, Dicotyledons; Kubitzki, K., Kadereit, J.W., Eds.; Springer: New York, NY, USA, 2004; Volume 7, pp. 296–306. [Google Scholar]
- Mitchell, R.J.; Beaton, J.K.; Bellamy, P.E.; Broome, A.; Chetcuti, J.; Eaton, S.; Ellis, C.J.; Gimona, A.; Harmer, R.; Hester, A.J.; et al. Ash dieback in the UK: A review of the ecological and conservation implications and potential management options. Biol. Conserv. 2014, 175, 95–109. [Google Scholar] [CrossRef]
- Wallander, E.; Albert, V.A. Phylogeny and classification of Oleaceae based on rps16 and trnL-F sequence data. Am. J. Bot. 2000, 87, 1827–1841. [Google Scholar] [CrossRef] [PubMed]
- Taylor, H. Cyto-taxonomy and phylogeny of the Oleaceae. Brittonia 1945, 5, 337–367. [Google Scholar] [CrossRef]
- Briggs, B.G. Some chromosome numbers in the Oleaceae. Contrib. N. S. W. Natl. Herb. 1970, 4, 126–129. [Google Scholar]
- George, K.; Geethamma, S. Cytology and evolution of jasmines. Cytologia 1992, 57, 27–32. [Google Scholar] [CrossRef][Green Version]
- Besnard, G.; Garcia-Verdugo, C.; Rubio de Casas, R.; Treier, U.A.; Galland, N.; Vargas, P. Polyploidy in the olive complex (Olea europaea): Evidence from flow cytometry and nuclear microsatellite analyses. Ann. Bot. 2008, 101, 25–30. [Google Scholar] [CrossRef]
- Lattier, J.D.; Contreras, R.N. Ploidy and genome size in lilac species, cultivars, and interploid hybrids. J. Am. Soc. Hortic. Sci. 2017, 142, 355–366. [Google Scholar] [CrossRef]
- Whittemore, A.T.; Cambell, J.J.N.; Zheng-Lian, X.; Carlson, C.H.; Atha, D.; Olsen, R.T. Ploidy variation in Fraxinus L. (Oleaceae) of eastern North America: Genome size diversity and taxonomy in a suddenly endangered genus. Int. J. Plant Sci. 2018, 179, 377–389. [Google Scholar] [CrossRef]
- Wallander, E. Systematics of Fraxinus (Oleaceae) and evolution of dioecy. Plant Syst. Evol. 2008, 273, 25–49. [Google Scholar] [CrossRef]
- Hinsinger, D.D.; Bask, J.; Gaudeul, M.; Cruaud, C.; Bertolino, P.; Frascaria-Lacoste, N.; Bousquet, J. The phylogeny and biogeographic history of ashes (Fraxinus, Oleaceae) highlight the roles of migration and vicariance in the diversification of temperate trees. PLoS ONE 2013, 8, e80431. [Google Scholar] [CrossRef]
- Rohwer, J.G. A preliminary survey of the fruits and seeds of the Oleaceae. Bot. Jahrb. Syst. 1993, 115, 271–291. [Google Scholar]
- Rohwer, J.G. Fruit and seed structures in Menodora (Oleaceae): A comparison with Jasminum. Bot. Acta 1995, 108, 163–168. [Google Scholar] [CrossRef]
- Rohwer, J.G. Die Frucht- und Samenstrukturen der Oleaceae. Bibl. Bot. 1996, 148, 1–177. [Google Scholar]
- Kiew, R. Preliminary pollen study of the Oleaceae in Malesia. Gard. Bull. 1984, 37, 225–230. [Google Scholar]
- Lepart, J.; Dommée, B. Is Phillyrea angustifolia L. (Oleaceae) an androdioecious species? Bot. J. Linn. Soc. 1992, 108, 375–387. [Google Scholar] [CrossRef]
- Green, P.S. A revision of Olea L. (Oleaceae). Kew Bull. 2002, 57, 91–140. [Google Scholar] [CrossRef]
- Saumitou-Laprade, P.; Vernet, P.; Dowkiw, A.; Bertrand, S.; Billiard, S.; Albert, B.; Gouyon, P.H.; Dufay, M. Polygamy or subdioecy? The impact of diallelic self-incompatibility on the sexual system in Fraxinus excelsior(Oleaceae). Proc. R. Soc. B Biol. Sci. 2018, 285, 20180004. [Google Scholar] [CrossRef]
- Thompson, J.D.; Dommée, B. Morph-specific patterns of variation in stigma height in natural populations of distylous Jasminum fruticans. New Phytol. 2000, 148, 303–314. [Google Scholar] [CrossRef]
- Olesen, J.M.; Dupont, Y.L.; Ehlers, B.K.; Valido, A.; Hansen, D.M. Heterostyly in the Canarian endemic Jasminum odoratissimurn (Oleaceae). Nord. J. Bot. 2005, 23, 537–539. [Google Scholar] [CrossRef]
- Ryu, T.Y.; Yeam, D.Y.; Kim, Y.J.; Kim, S.J. Studies on heterostyly incompatibility of Abeliophyllum distichum. Seoul Natl. Univ. Coll. Agric. Bull. 1976, 1, 113–120. [Google Scholar]
- Saumitou-Laprade, P.; Vernet, P.; Vassiliadis, C.; Hoareau, Y.; de Magny, G.; Dommée, B.; Lepart, J. A self-incompatibility system explains high male frequencies in an androdioecious plant. Science 2010, 327, 1648–1650. [Google Scholar] [CrossRef] [PubMed]
- Vernet, P.; Lepercq, P.; Billiard, S.; Bourceaux, A.; Lepart, J.; Dommée, B.; Saumitou-Laprade, P. Evidence for the long-term maintenance of a rare self-incompatibility system in Oleaceae. New Phytol. 2016, 210, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.S. A review of the family Oleaceae. Contrib. N. S. W. Natl. Herb. 1957, 2, 395–418. [Google Scholar]
- Stearn, W.T. Union of Chionanthus and Linociera (Oleaceae). Ann. Mo. Bot. Gard. 1976, 63, 355–357. [Google Scholar] [CrossRef]
- Li, J.; Alexander, J.H.; Zhang, D. Paraphyletic Syringa (Oleaceae): Evidence from sequences of nuclear ribosomal DNA ITS and ETS regions. Syst. Bot. 2002, 27, 592–597. [Google Scholar]
- Harborne, J.B.; Green, P.S. A chemotaxonomic survey of flvonoids in leaves of the Oleaceae. Bot. J. Linn. Soc. 1980, 81, 155–167. [Google Scholar] [CrossRef]
- Besnard, G.; Rubio de Casas, R.; Christin, P.A.; Vargas, P. Phylogenetics of Olea (Oleaceae) based on plastid and nuclear ribosomal DNA sequences: Tertiary climatic shifts and lineage differentiation times. Ann. Bot. 2009, 104, 143–160. [Google Scholar] [CrossRef]
- Yuan, W.J.; Zhang, W.R.; Han, Y.J.; Dong, M.F.; Shang, F.D. Molecular phylogeny of Osmanthus (Oleaceae) based on non-coding chloroplast and nuclear ribosomal internal transcribed spacer regions. J. Syst. Evol. 2010, 48, 482–489. [Google Scholar] [CrossRef]
- Hong-Wa, C.; Besnard, G. Intricate patterns of phylogenetic relationships in the olive family as inferred from multi-locus plastid and nuclear DNA sequence analyses: A close-up on Chionanthus and Noronhia (Oleaceae). Mol. Phylogenet. Evol. 2013, 67, 367–378. [Google Scholar] [CrossRef]
- Olofsson, J.K.; Cantera, I.; Van de Paer, C.; Hong-Wa, C.; Zedane, L.; Dunning, L.T.; Alberti, A.; Christin, P.A.; Besnard, G. Phylogenomics using low-depth whole genome sequencing: A case study with the olive tribe. Mol. Ecol. Resour. 2019, 19, 877–892. [Google Scholar] [CrossRef]
- Li, Y.F.; Zhang, M.; Wang, X.R.; Sylvester, S.P.; Xiang, Q.B.; Li, X.; Li, M.; Zhu, H.; Zhang, C.; Chen, L.; et al. Revisiting the phylogeny and taxonomy of Osmanthus (Oleaceae) including description of the new genus Chengiodendron. Phytotaxa 2020, 436, 283–292. [Google Scholar] [CrossRef]
- Kim, K.J.; Jansen, R.K. A chloroplast DNA phylogeny of lilacs (Syringa, Oleaceae): Plastome groups show a strong correlation with crossing groups. Am. J. Bot. 1998, 85, 1338–1351. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.L.; Jansen, R.K.; Chumley, T.W.; Kim, K.J. Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 2007, 24, 1161–1180. [Google Scholar] [CrossRef]
- Kim, D.; Kim, J. Molecular phylogeny of tribe Forsythieae (Oleaceae) based on nuclear ribosomal DNA internal transcribed spacers and plastid DNA trnL-F and matK gene sequences. J. Plant Res. 2011, 124, 339–347. [Google Scholar] [CrossRef]
- Ha, Y.H.; Kim, C.; Choi, K.; Kim, J.H. Molecular phylogeny and dating of Forsythieae (Oleaceae) provide insight into the Miocene history of Eurasian temperate shrubs. Front. Plant Sci. 2018, 9, 99. [Google Scholar] [CrossRef] [PubMed]
- Jeyarani, J.N.; Yohannan, R.; Vijayavalli, D.; Dwivedi, M.D.; Pandey, A.K. Phylogenetic analysis and evolution of morphological characters in the genus Jasminum L. (Oleaceae) in India. J. Genet. 2018, 97, 1225–1239. [Google Scholar] [CrossRef]
- Cruz, F.; Julca, I.; Gómez-Garrido, J.; Loska, D.; Marcet-Houben, M.; Cano, E.; Galán, B.; Frias, L.; Ribeca, P.; Derdak, S.; et al. Genome sequence of the olive tree, Olea europaea. GigaScience 2016, 5, 29. [Google Scholar] [CrossRef]
- Unver, T.; Wu, Z.; Sterck, L.; Turktas, M.; Lohaus, R.; Li, Z.; Yang, M.; He, L.; Deng, T.; Escalante, F.J.; et al. Genome of wild olive and the evolution of oil biosynthesis. Proc. Natl. Acad. Sci. USA 2017, 114, E9413–E9422. [Google Scholar] [CrossRef]
- Sollars, E.S.A.; Harper, A.L.; Kelly, L.J.; Sambles, C.M.; Ramirez-Gonzalez, R.H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E.D.; Uauy, C.; Havlickova, L.; et al. Genome sequence and genetic diversity of European ash trees. Nature 2017, 541, 212–216. [Google Scholar] [CrossRef]
- Kelly, L.J.; Plumb, W.J.; Carey, D.W.; Mason, M.E.; Cooper, E.D.; Crowther, W.; Whittemore, A.T.; Rossiter, S.J.; Kock, J.L.; Buggs, R.J.A. Convergent molecular evolution among ash species resistant to the emerald ash borer. Nat. Ecol. Evol. 2020, 4, 1116–1128. [Google Scholar] [CrossRef]
- Van de Paer, C.; Bouchez, O.; Besnard, G. Prospects on the evolutionary mitogenomics of plants: A case study on the olive family (Oleaceae). Mol. Ecol. Resour. 2018, 18, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, T.; Luebert, F.; Xiang, Y.; Huang, C.H.; Hu, Y.; Rees, M.; Frohlich, M.W.; Qi, J.; Weigend, M.; et al. Asterid phylogenomics/phylotranscriptomics uncover morphological evolutionary histories and support phylogenetic placement for numerous whole-genome duplications. Mol. Biol. Evol. 2020, 37, 3188–3210. [Google Scholar] [CrossRef] [PubMed]
- Bieker, V.C.; Martin, M.D. Implications and future prospects for evolutionary analyses of DNA in historical herbarium collections. Bot. Lett. 2018, 165, 409–418. [Google Scholar] [CrossRef]
- Van de Paer, C.; Hong-Wa, C.; Jeziorski, C.; Besnard, G. Mitogenomics of Hesperelaea, an extinct genus of Oleaceae. Gene 2016, 594, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Zedane, L.; Hong-Wa, C.; Murienne, J.; Jeziorski, C.; Baldwin, B.G.; Besnard, G. Museomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Biol. J. Linn. Soc. 2016, 117, 44–57. [Google Scholar] [CrossRef]
- Straub, S.C.K.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef]
- Berger, B.A.; Han, J.; Sessa, E.B.; Gardner, A.G.; Shepherd, K.A.; Ricigliano, V.A.; Jabaily, R.S.; Howarth, D.G. The unexpected depths of genome-skimming data: A case study examining Goodeniaceae floral symmetry genes. Appl. Plant Sci. 2017, 5, 1700042. [Google Scholar] [CrossRef]
- Govindarajulu, R.; Parks, M.; Tennessen, J.A.; Liston, A.; Ashman, T.L. Comparison of nuclear, plastid, and mitochondrial phylogenies and the origin of wild octoploid strawberry species. Am. J. Bot. 2015, 102, 544–554. [Google Scholar] [CrossRef]
- Sun, M.; Soltis, D.E.; Soltis, P.S.; Zhu, X.; Burleigh, J.G.; Chen, Z. Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenet. Evol. 2015, 83, 156–166. [Google Scholar] [CrossRef]
- Liu, S.H.; Edwards, C.E.; Hoch, P.C.; Raven, P.H.; Barber, J.C. Genome skimming provides new insight into the relationships in Ludwigia section Macrocarpon, a polyploid complex. Am. J. Bot. 2018, 105, 875–887. [Google Scholar] [CrossRef]
- Mathews, S.; Lavin, M.; Sharrock, R.A. Evolution of the phytochrome gene family and its utility for phylogenetic analyses of angiosperms. Ann. Mo. Bot. Gard. 1995, 82, 296–321. [Google Scholar] [CrossRef]
- Mathews, S.; Tsai, R.C.; Kellogg, E.A. Phylogenetic structure in the grass family (Poaceae): Evidence from the nuclear gene phytochrome B. Am. J. Bot. 2000, 87, 96–107. [Google Scholar] [CrossRef] [PubMed]
- WCSP. World Checklist of Selected Plant Families. Facilitated by the Royal Botanic Gardens, Kew. 2020. Published on the Internet. Available online: http://wcsp.science.kew.org/ (accessed on 4 June 2020).
- Banfi, E. Chrysojasminum, a new genus for Jasminum sect. Alternifolia (Oleaceae, Jasmineae). Nat. Hist. Sci. 2014, 1, 3–6. [Google Scholar] [CrossRef]
- Bianconi, M.; Hackel, J.; Vorontsova, M.S.; Alberti, A.; Arthan, W.; Burke, S.V.; Duvall, M.R.; Kellogg, E.A.; Lavergne, S.; McKain, M.; et al. Continued adaptation of C4 photosynthesis after an initial burst of changes in the Andropogoneae grasses. Syst. Biol. 2020, 69, 445–461. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. GENEIOUS Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Meth. 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Besnard, G.; Christin, P.A.; Malé, P.J.; Coissac, E.; Lhuillier, E.; Lauzeral, C.; Vorontsova, M.S. From museums to genomics: Old herbarium specimens shed light on a C3 to C4 transition. J. Exp. Bot. 2014, 65, 6711–6721. [Google Scholar] [CrossRef]
- Besnard, G.; Bianconi, M.E.; Hackel, J.; Manzi, S.; Vorontsova, M.S.; Christin, P.A. Herbarium genomics retrace the origins of C4-specific carbonic anhydrase in Andropogoneae (Poaceae). Bot. Lett. 2018, 165, 419–433. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Löytynoja, A. Phylogeny-aware alignment with PRANK. Meth. Mol. Biol. 2014, 1079, 155–170. [Google Scholar]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Meth. 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef]
- Phillips, M.J.; Delsuc, F.; Penny, D. Genome-scale phylogeny and the detection of systematic biases. Mol. Biol. Evol. 2004, 21, 1455–1458. [Google Scholar] [CrossRef]
- Minh, B.Q.; Hahn, M.W.; Lanfear, R. New methods to calculate concordance factors for phylogenomic datasets. Mol. Biol. Evol. 2020, 37, 2727–2733. [Google Scholar] [CrossRef]
- Kiew, R.; Baas, P. Nyctanthes is a member of Oleaceae. Proc. Ind. Acad. Sci. 1984, 93, 349–358. [Google Scholar]
- George, K.; Geethamma, S. Cytological and other evidences for the taxonomic position of Nyctanthes arbor-tristis. Curr. Sci. 1984, 53, 439–441. [Google Scholar]
- Huang, C.H.; Zhang, C.; Liu, M.; Hu, Y.; Gao, T.; Qi, J.; Ma, H. Multiple polyploidization events across Asteraceae with two nested events in the early history revealed by nuclear phylogenomics. Mol. Biol. Evol. 2016, 33, 2820–2835. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ding, Y.; Zhu, C.D.; Zhou, X.; Orr, M.C.; Scheu, S.; Luan, Y.X. Phylogenomics from low-coverage whole-genome sequencing. Meth. Ecol. Evol. 2019, 10, 507–517. [Google Scholar] [CrossRef]
- Smith, S.A.; Donoghue, M.J. Rates of molecular evolution are linked to life history in flowering plants. Science 2008, 322, 86–89. [Google Scholar] [CrossRef]
- Larracuente, A.M.; Sackton, T.B.; Greenberg, A.J.; Wong, A.; Singh, N.D.; Sturgill, D.; Zhang, Y.; Oliver, B.; Clark, A.G. Evolution of protein-coding genes in Drosophila. Trends Genet. 2008, 24, 114–123. [Google Scholar] [CrossRef]
- Yang, L.; Gaut, B.S. Factors that contribute to variation in evolutionary rate among Arabidopsis genes. Mol. Biol. Evol. 2011, 28, 2359–2369. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupin, J.; Raimondeau, P.; Hong-Wa, C.; Manzi, S.; Gaudeul, M.; Besnard, G. Resolving the Phylogeny of the Olive Family (Oleaceae): Confronting Information from Organellar and Nuclear Genomes. Genes 2020, 11, 1508. https://doi.org/10.3390/genes11121508
Dupin J, Raimondeau P, Hong-Wa C, Manzi S, Gaudeul M, Besnard G. Resolving the Phylogeny of the Olive Family (Oleaceae): Confronting Information from Organellar and Nuclear Genomes. Genes. 2020; 11(12):1508. https://doi.org/10.3390/genes11121508
Chicago/Turabian StyleDupin, Julia, Pauline Raimondeau, Cynthia Hong-Wa, Sophie Manzi, Myriam Gaudeul, and Guillaume Besnard. 2020. "Resolving the Phylogeny of the Olive Family (Oleaceae): Confronting Information from Organellar and Nuclear Genomes" Genes 11, no. 12: 1508. https://doi.org/10.3390/genes11121508
APA StyleDupin, J., Raimondeau, P., Hong-Wa, C., Manzi, S., Gaudeul, M., & Besnard, G. (2020). Resolving the Phylogeny of the Olive Family (Oleaceae): Confronting Information from Organellar and Nuclear Genomes. Genes, 11(12), 1508. https://doi.org/10.3390/genes11121508