Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones


 and
and 
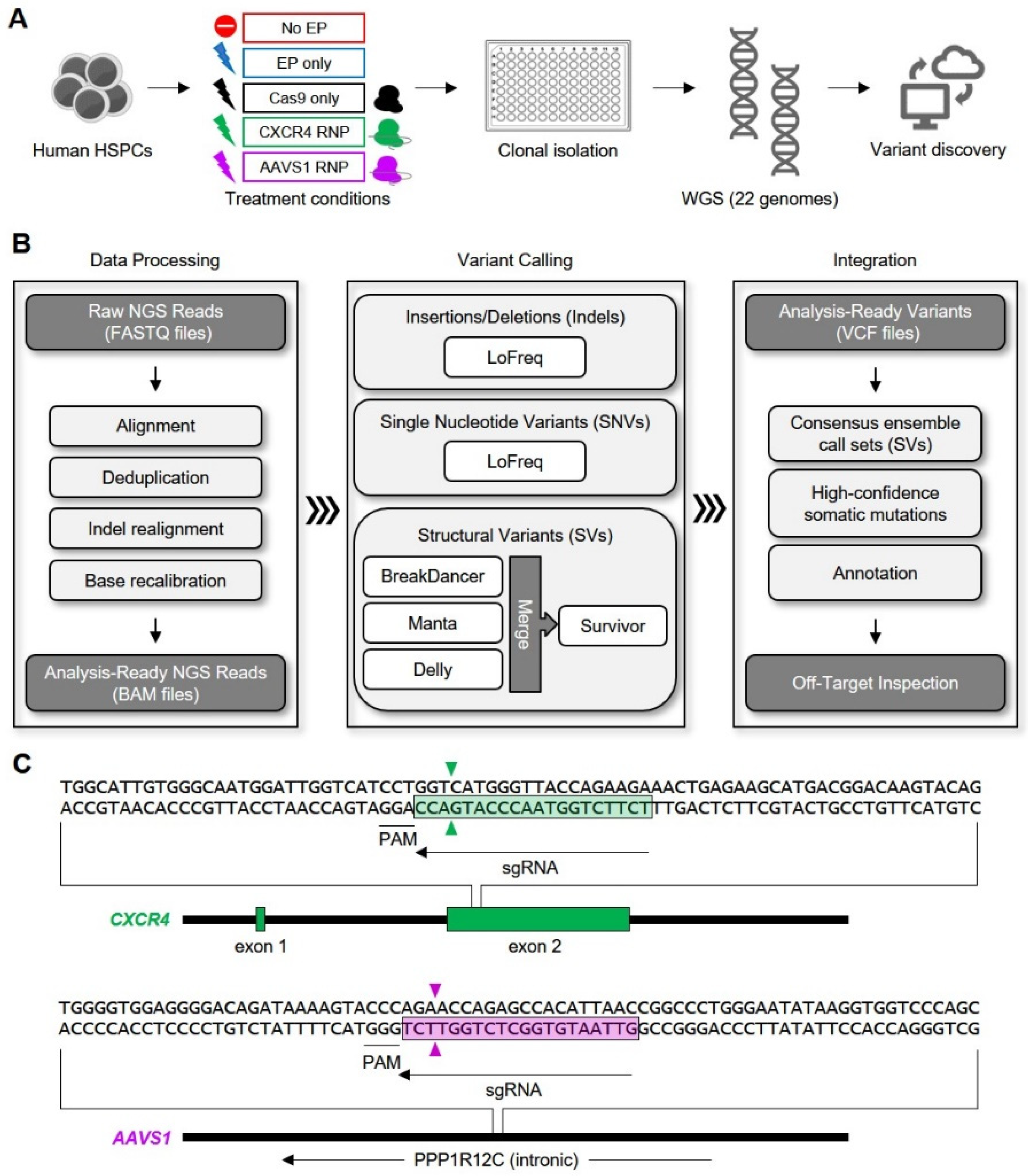{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Human HSPC Isolation
2.2. Electroporation of Human HSPCs
2.3. Single-Cell Cloning
2.4. DNA Isolation
2.5. T7 Endonuclease I Assay
2.6. NGS Library Preparation and High-Throughput Illumina Sequencing
2.7. Bioinformatic Analysis
2.8. Data Availability
2.9. Statistical Analysis
3. Results
3.1. Genome-Wide Analysis of Cas9 Promiscuity in Single-Cell-Derived Human HSPC Clones
3.2. Characterization of Indels and SNVs
3.3. Characterization of Structural Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cavazzana, M.; Bushman, F.D.; Miccio, A.; Andre-Schmutz, I.; Six, E. Gene therapy targeting haematopoietic stem cells for inherited diseases: Progress and challenges. Nat. Rev. Drug Discov. 2019, 18, 447–462. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Bak, R.O.; Gomez-Ospina, N.; Porteus, M.H. Gene Editing on Center Stage. Trends Genet. 2018, 34, 600–611. [Google Scholar] [CrossRef]
- Porteus, M.H. A New Class of Medicines through DNA Editing. N. Engl. J. Med. 2019, 380, 947–959. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef]
- Rousseau, B.A.; Hou, Z.; Gramelspacher, M.J.; Zhang, Y. Programmable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 System from Neisseria meningitidis. Mol. Cell 2018, 69, 906–914.e904. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Cui, Y.; Xu, J.; Cheng, M.; Liao, X.; Peng, S. Review of CRISPR/Cas9 sgRNA Design Tools. Interdiscip. Sci. 2018, 10, 455–465. [Google Scholar] [CrossRef]
- Wilson, L.O.W.; O’Brien, A.R.; Bauer, D.C. The Current State and Future of CRISPR-Cas9 gRNA Design Tools. Front. Pharm. 2018, 9, 749. [Google Scholar] [CrossRef]
- Hu, J.; Meyers, R.M.; Dong, J.; Panchakshari, R.A.; Alt, F.W.; Frock, R.L. Detecting DNA double-stranded breaks in mammalian genomes by linear amplification-mediated high-throughput genome-wide translocation sequencing. Nat. Protoc. 2016, 11, 853–871. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.J.; Yee, J.K. Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef]
- Akcakaya, P.; Bobbin, M.L.; Guo, J.A.; Malagon-Lopez, J.; Clement, K.; Garcia, S.P.; Fellows, M.D.; Porritt, M.J.; Firth, M.A.; Carreras, A.; et al. In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 2018, 561, 416–419. [Google Scholar] [CrossRef]
- Wienert, B.; Wyman, S.K.; Richardson, C.D.; Yeh, C.D.; Akcakaya, P.; Porritt, M.J.; Morlock, M.; Vu, J.T.; Kazane, K.R.; Watry, H.L.; et al. Unbiased detection of CRISPR off-targets in vivo using DISCOVER-Seq. Science 2019, 364, 286–289. [Google Scholar] [CrossRef]
- Yan, W.X.; Mirzazadeh, R.; Garnerone, S.; Scott, D.; Schneider, M.W.; Kallas, T.; Custodio, J.; Wernersson, E.; Li, Y.; Gao, L.; et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun. 2017, 8, 15058. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef]
- Tsai, S.Q.; Nguyen, N.T.; Malagon-Lopez, J.; Topkar, V.V.; Aryee, M.J.; Joung, J.K. CIRCLE-seq: A highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 2017, 14, 607–614. [Google Scholar] [CrossRef]
- Kim, D.; Bae, S.; Park, J.; Kim, E.; Kim, S.; Yu, H.R.; Hwang, J.; Kim, J.I.; Kim, J.S. Digenome-seq: Genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat. Methods 2015, 12, 237–243. [Google Scholar] [CrossRef]
- Cameron, P.; Fuller, C.K.; Donohoue, P.D.; Jones, B.N.; Thompson, M.S.; Carter, M.M.; Gradia, S.; Vidal, B.; Garner, E.; Slorach, E.M.; et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods 2017, 14, 600–606. [Google Scholar] [CrossRef]
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404. [Google Scholar] [CrossRef]
- Kim, D.; Kim, S.; Kim, S.; Park, J.; Kim, J.S. Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-seq. Genome Res. 2016, 26, 406–415. [Google Scholar] [CrossRef]
- Smith, C.; Gore, A.; Yan, W.; Abalde-Atristain, L.; Li, Z.; He, C.; Wang, Y.; Brodsky, R.A.; Zhang, K.; Cheng, L.; et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell 2014, 15, 12–13. [Google Scholar] [CrossRef]
- Suzuki, K.; Yu, C.; Qu, J.; Li, M.; Yao, X.; Yuan, T.; Goebl, A.; Tang, S.; Ren, R.; Aizawa, E.; et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 2014, 15, 31–36. [Google Scholar] [CrossRef]
- Tang, L.; Zeng, Y.; Du, H.; Gong, M.; Peng, J.; Zhang, B.; Lei, M.; Zhao, F.; Wang, W.; Li, X.; et al. CRISPR/Cas9-mediated gene editing in human zygotes using Cas9 protein. Mol. Genet. Genom. 2017, 292, 525–533. [Google Scholar] [CrossRef]
- Veres, A.; Gosis, B.S.; Ding, Q.; Collins, R.; Ragavendran, A.; Brand, H.; Erdin, S.; Cowan, C.A.; Talkowski, M.E.; Musunuru, K. Low incidence of off-target mutations in individual CRISPR-Cas9 and TALEN targeted human stem cell clones detected by whole-genome sequencing. Cell Stem Cell 2014, 15, 27–30. [Google Scholar] [CrossRef]
- Anderson, K.R.; Haeussler, M.; Watanabe, C.; Janakiraman, V.; Lund, J.; Modrusan, Z.; Stinson, J.; Bei, Q.; Buechler, A.; Yu, C.; et al. CRISPR off-target analysis in genetically engineered rats and mice. Nat. Methods 2018, 15, 512–514. [Google Scholar] [CrossRef]
- Dong, Y.; Li, H.; Zhao, L.; Koopman, P.; Zhang, F.; Huang, J.X. Genome-Wide Off-Target Analysis in CRISPR-Cas9 Modified Mice and Their Offspring. G3 Genes Genomes Genet. 2019, 9, 3645–3651. [Google Scholar] [CrossRef]
- Iyer, V.; Boroviak, K.; Thomas, M.; Doe, B.; Riva, L.; Ryder, E.; Adams, D.J. No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 2018, 14, e1007503. [Google Scholar] [CrossRef]
- Iyer, V.; Shen, B.; Zhang, W.; Hodgkins, A.; Keane, T.; Huang, X.; Skarnes, W.C. Off-target mutations are rare in Cas9-modified mice. Nat. Methods 2015, 12, 479. [Google Scholar] [CrossRef]
- Kim, S.T.; Park, J.; Kim, D.; Kim, K.; Bae, S.; Schlesner, M.; Kim, J.S. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 2018, 15, 239–240. [Google Scholar] [CrossRef]
- Lareau, C.A.; Clement, K.; Hsu, J.Y.; Pattanayak, V.; Joung, J.K.; Aryee, M.J.; Pinello, L. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 2018, 15, 238–239. [Google Scholar] [CrossRef]
- Lescarbeau, R.M.; Murray, B.; Barnes, T.M.; Bermingham, N. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 2018, 15, 237. [Google Scholar] [CrossRef]
- Willi, M.; Smith, H.E.; Wang, C.; Liu, C.; Hennighausen, L. Mutation frequency is not increased in CRISPR-Cas9-edited mice. Nat. Methods 2018, 15, 756–758. [Google Scholar] [CrossRef]
- Wilson, C.J.; Fennell, T.; Bothmer, A.; Maeder, M.L.; Reyon, D.; Cotta-Ramusino, C.; Fernandez, C.A.; Marco, E.; Barrera, L.A.; Jayaram, H.; et al. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 2018, 15, 236–237. [Google Scholar] [CrossRef]
- Li, C.; Zhou, S.; Li, Y.; Li, G.; Ding, Y.; Li, L.; Liu, J.; Qu, L.; Sonstegard, T.; Huang, X.; et al. Trio-Based Deep Sequencing Reveals a Low Incidence of Off-Target Mutations in the Offspring of Genetically Edited Goats. Front. Genet. 2018, 9, 449. [Google Scholar] [CrossRef]
- Li, J.; Manghwar, H.; Sun, L.; Wang, P.; Wang, G.; Sheng, H.; Zhang, J.; Liu, H.; Qin, L.; Rui, H.; et al. Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR/Cas9-edited cotton plants. Plant Biotechnol. J. 2019, 17, 858–868. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef]
- Cradick, T.J.; Fine, E.J.; Antico, C.J.; Bao, G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013, 41, 9584–9592. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef]
- Morgens, D.W.; Wainberg, M.; Boyle, E.A.; Ursu, O.; Araya, C.L.; Tsui, C.K.; Haney, M.S.; Hess, G.T.; Han, K.; Jeng, E.E.; et al. Genome-scale measurement of off-target activity using Cas9 toxicity in high-throughput screens. Nat. Commun. 2017, 8, 15178. [Google Scholar] [CrossRef]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Hendel, A.; Bak, R.O.; Clark, J.T.; Kennedy, A.B.; Ryan, D.E.; Roy, S.; Steinfeld, I.; Lunstad, B.D.; Kaiser, R.J.; Wilkens, A.B.; et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef]
- Ryan, D.E.; Taussig, D.; Steinfeld, I.; Phadnis, S.M.; Lunstad, B.D.; Singh, M.; Vuong, X.; Okochi, K.D.; McCaffrey, R.; Olesiak, M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. [Google Scholar] [CrossRef]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef]
- Chen, J.S.; Dagdas, Y.S.; Kleinstiver, B.P.; Welch, M.M.; Sousa, A.A.; Harrington, L.B.; Sternberg, S.H.; Joung, J.K.; Yildiz, A.; Doudna, J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 2017, 550, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kulcsar, P.I.; Talas, A.; Huszar, K.; Ligeti, Z.; Toth, E.; Weinhardt, N.; Fodor, E.; Welker, E. Crossing enhanced and high fidelity SpCas9 nucleases to optimize specificity and cleavage. Genome Biol. 2017, 18, 190. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Jeong, E.; Lee, J.; Jung, M.; Shin, E.; Kim, Y.H.; Lee, K.; Jung, I.; Kim, D.; Kim, S.; et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat. Commun. 2018, 9, 3048. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Zook, J.M.; Samarov, D.; McDaniel, J.; Sen, S.K.; Salit, M. Synthetic spike-in standards improve run-specific systematic error analysis for DNA and RNA sequencing. PLoS ONE 2012, 7, e41356. [Google Scholar] [CrossRef]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wallis, J.W.; McLellan, M.D.; Larson, D.E.; Kalicki, J.M.; Pohl, C.S.; McGrath, S.D.; Wendl, M.C.; Zhang, Q.; Locke, D.P.; et al. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat. Methods 2009, 6, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Kallberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Rausch, T.; Zichner, T.; Schlattl, A.; Stutz, A.M.; Benes, V.; Korbel, J.O. DELLY: Structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012, 28, i333–i339. [Google Scholar] [CrossRef] [PubMed]
- Jeffares, D.C.; Jolly, C.; Hoti, M.; Speed, D.; Shaw, L.; Rallis, C.; Balloux, F.; Dessimoz, C.; Bahler, J.; Sedlazeck, F.J. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017, 8, 14061. [Google Scholar] [CrossRef] [PubMed]
- Papapetrou, E.P.; Schambach, A. Gene Insertion Into Genomic Safe Harbors for Human Gene Therapy. Mol. Ther. 2016, 24, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.; Park, J.; Kim, J.S. Cas-OFFinder: A fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 2014, 30, 1473–1475. [Google Scholar] [CrossRef]
- Hay, E.A.; Khalaf, A.R.; Marini, P.; Brown, A.; Heath, K.; Sheppard, D.; MacKenzie, A. An analysis of possible off target effects following CAS9/CRISPR targeted deletions of neuropeptide gene enhancers from the mouse genome. Neuropeptides 2017, 64, 101–107. [Google Scholar] [CrossRef]
- Nakajima, K.; Kazuno, A.A.; Kelsoe, J.; Nakanishi, M.; Takumi, T.; Kato, T. Exome sequencing in the knockin mice generated using the CRISPR/Cas system. Sci. Rep. 2016, 6, 34703. [Google Scholar] [CrossRef]
- Nutter, L.M.J.; Heaney, J.D.; Lloyd, K.C.K.; Murray, S.A.; Seavitt, J.R.; Skarnes, W.C.; Teboul, L.; Brown, S.D.M.; Moore, M. Response to “Unexpected mutations after CRISPR-Cas9 editing in vivo”. Nat. Methods 2018, 15, 235–236. [Google Scholar] [CrossRef]
- Varshney, G.K.; Pei, W.; LaFave, M.C.; Idol, J.; Xu, L.; Gallardo, V.; Carrington, B.; Bishop, K.; Jones, M.; Li, M.; et al. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res. 2015, 25, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Ferreira, L.M.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Newton, M.D.; Taylor, B.J.; Driessen, R.P.C.; Roos, L.; Cvetesic, N.; Allyjaun, S.; Lenhard, B.; Cuomo, M.E.; Rueda, D.S. DNA stretching induces Cas9 off-target activity. Nat. Struct. Mol. Biol. 2019, 26, 185–192. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, R.H.; Chen, Y.-C.; Seifuddin, F.; Hupalo, D.; Alba, C.; Reger, R.; Tian, X.; Araki, D.; Dalgard, C.L.; Childs, R.W.; et al. Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones. Genes 2020, 11, 1501. https://doi.org/10.3390/genes11121501
Smith RH, Chen Y-C, Seifuddin F, Hupalo D, Alba C, Reger R, Tian X, Araki D, Dalgard CL, Childs RW, et al. Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones. Genes. 2020; 11(12):1501. https://doi.org/10.3390/genes11121501
Chicago/Turabian StyleSmith, Richard H., Yun-Ching Chen, Fayaz Seifuddin, Daniel Hupalo, Camille Alba, Robert Reger, Xin Tian, Daisuke Araki, Clifton L. Dalgard, Richard W. Childs, and et al. 2020. "Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones" Genes 11, no. 12: 1501. https://doi.org/10.3390/genes11121501
APA StyleSmith, R. H., Chen, Y.-C., Seifuddin, F., Hupalo, D., Alba, C., Reger, R., Tian, X., Araki, D., Dalgard, C. L., Childs, R. W., Pirooznia, M., & Larochelle, A. (2020). Genome-Wide Analysis of Off-Target CRISPR/Cas9 Activity in Single-Cell-Derived Human Hematopoietic Stem and Progenitor Cell Clones. Genes, 11(12), 1501. https://doi.org/10.3390/genes11121501