Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic

 , , , ,
, , , ,  , and add
Show full author list
, and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Eligibility Criteria
2.2. Histological Assessment
2.3. Sample Preparation and DNA Extraction
2.4. Clinicopathological Analysis
2.5. Library Preparation
2.6. Bioinformatic Analysis
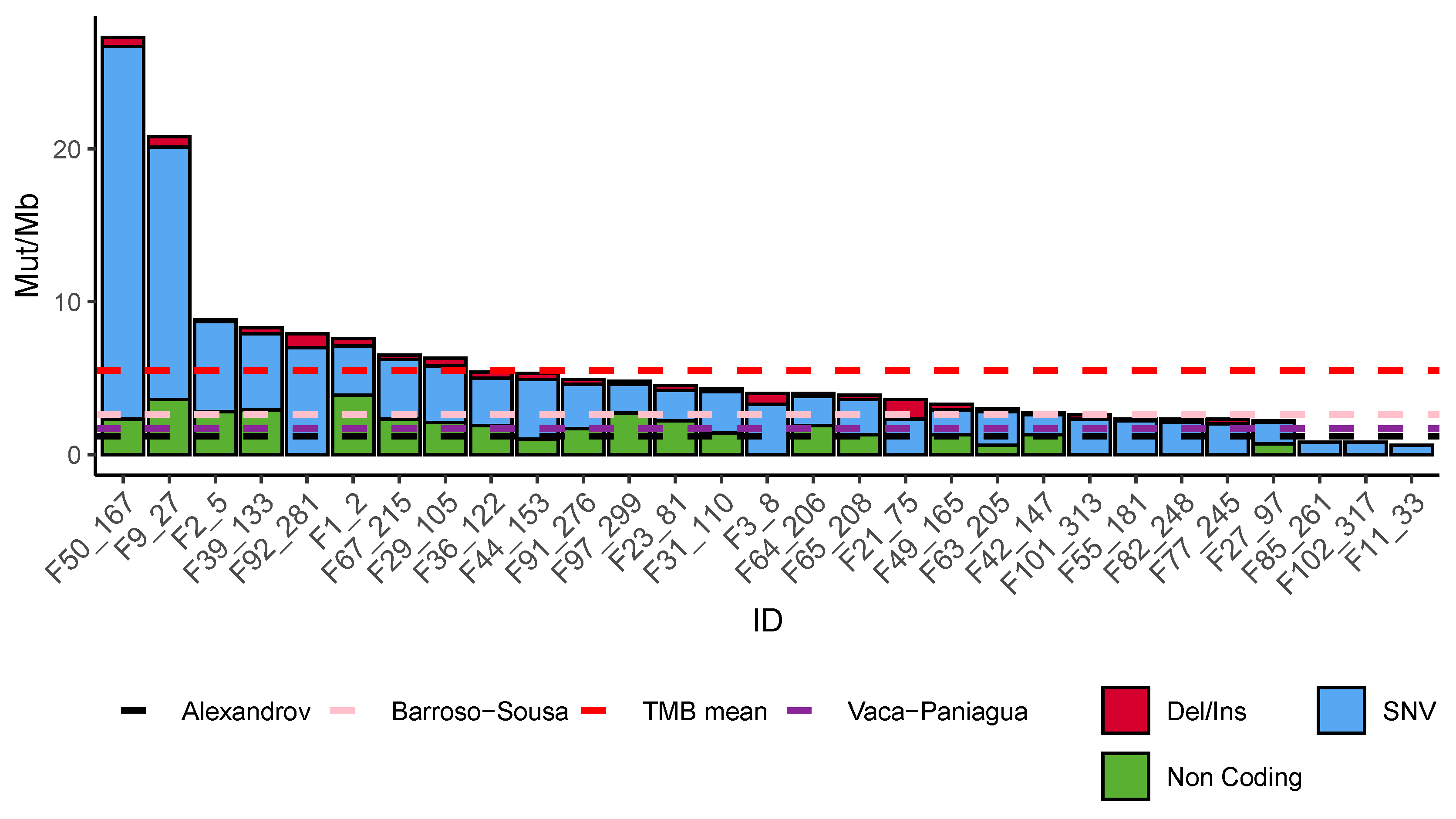
2.7. Tumor Mutational Burden Analysis
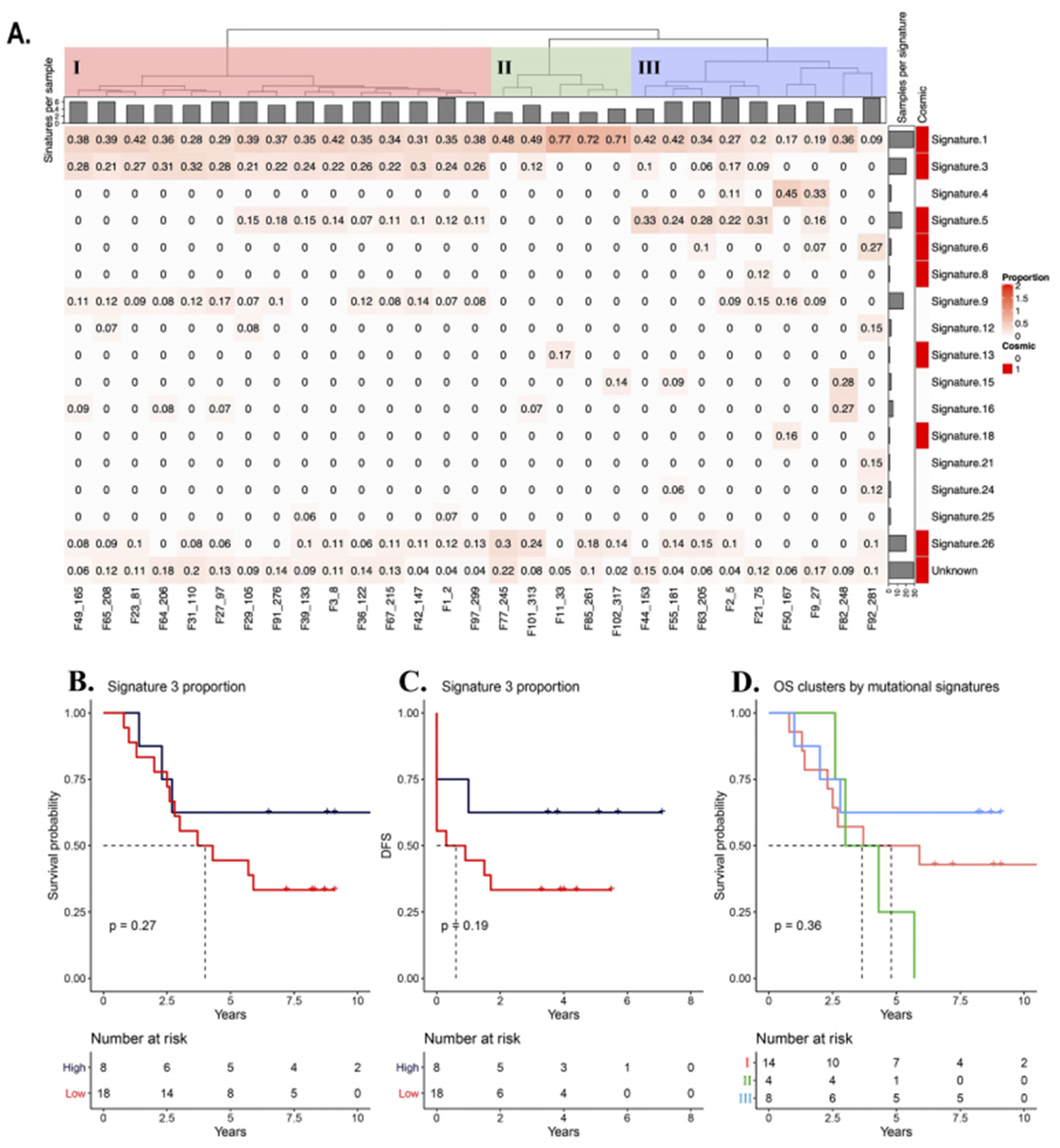
2.8. Mutational Signature Analysis
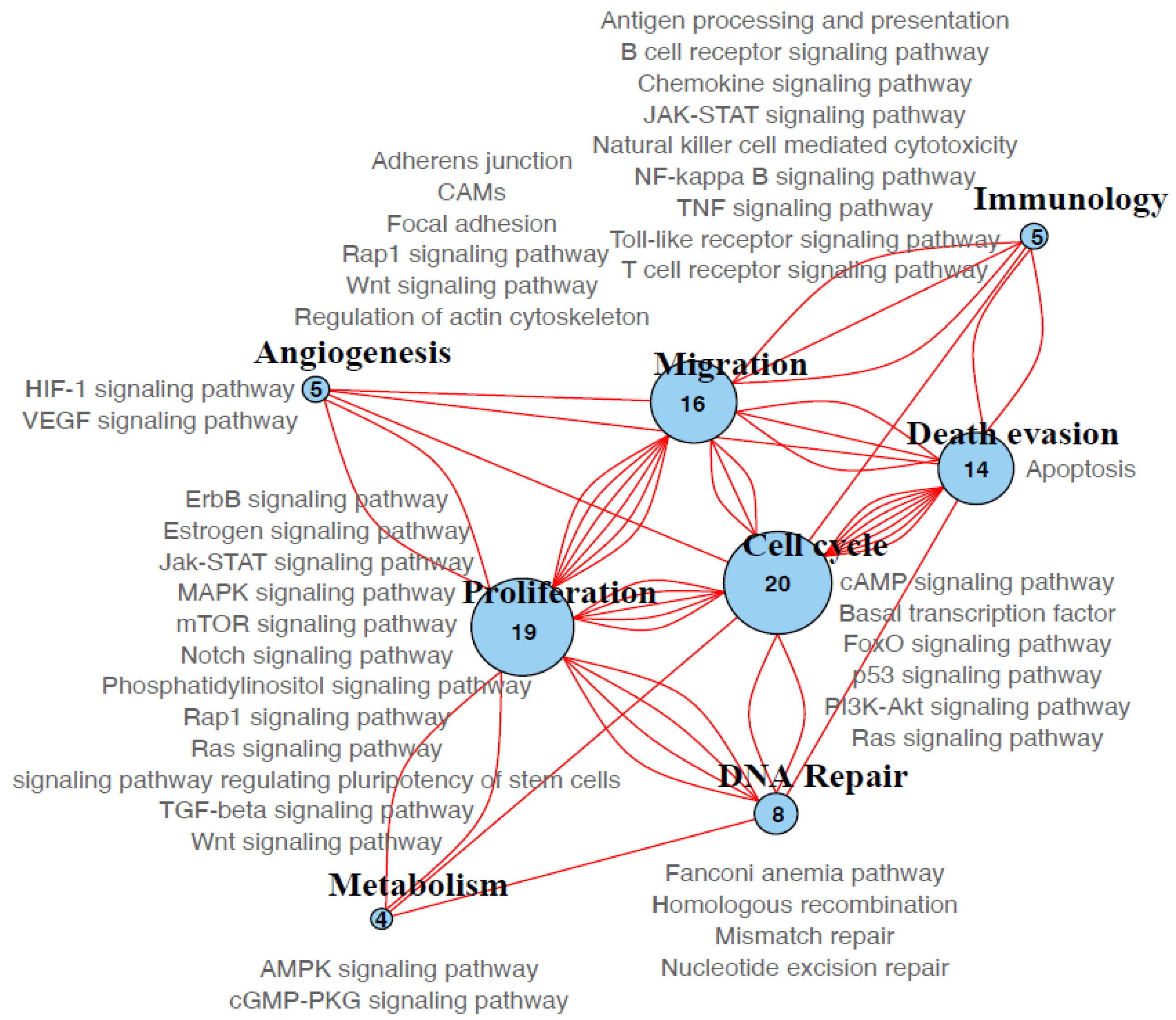
2.9. Pathway Enrichment Analysis
2.10. Statistical and Survival Analysis
2.11. Global Actionable Alterations
3. Results
3.1. Patient and Tumor Characteristics
3.2. Overall Survival and Complete Pathological Response
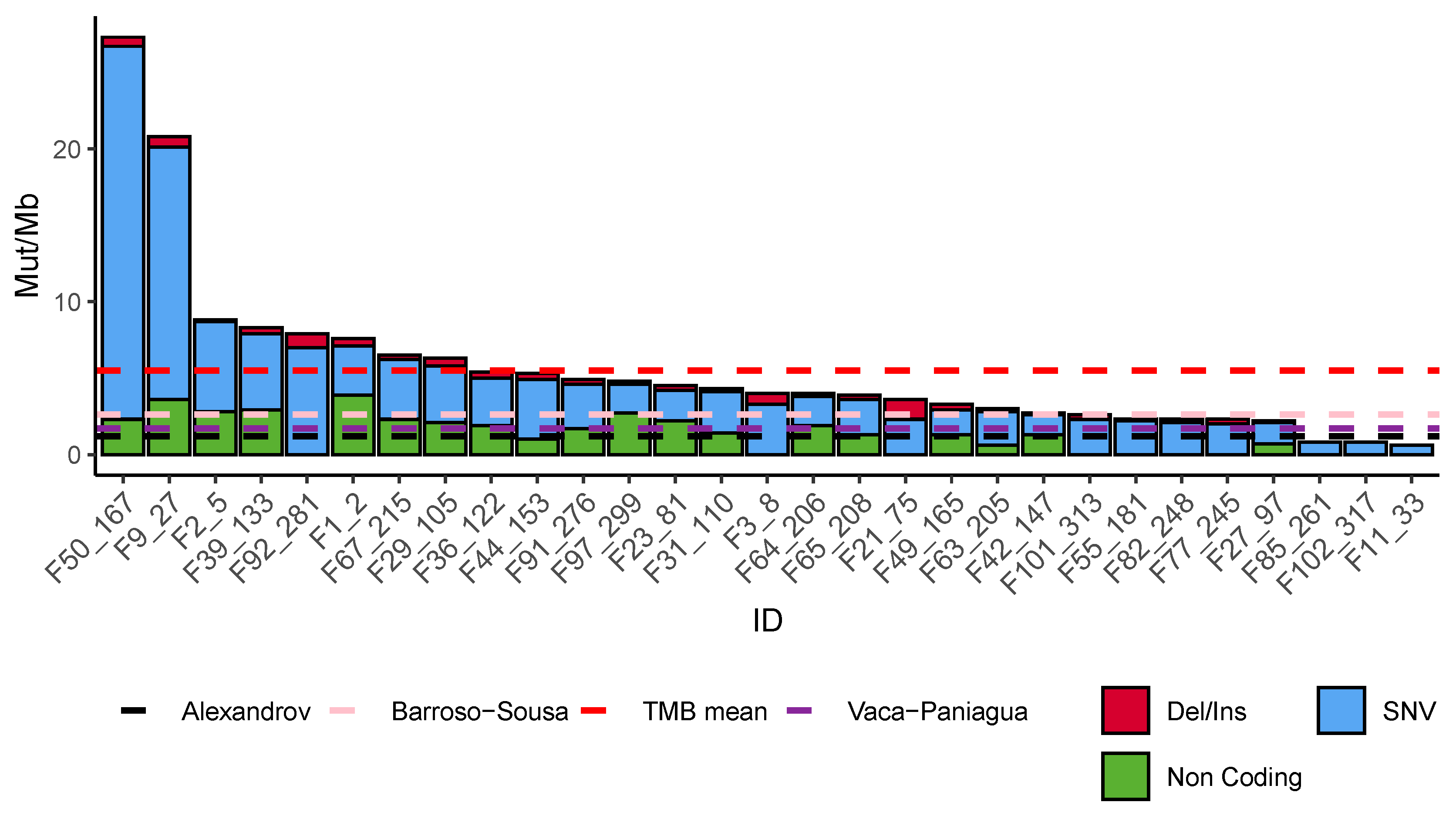
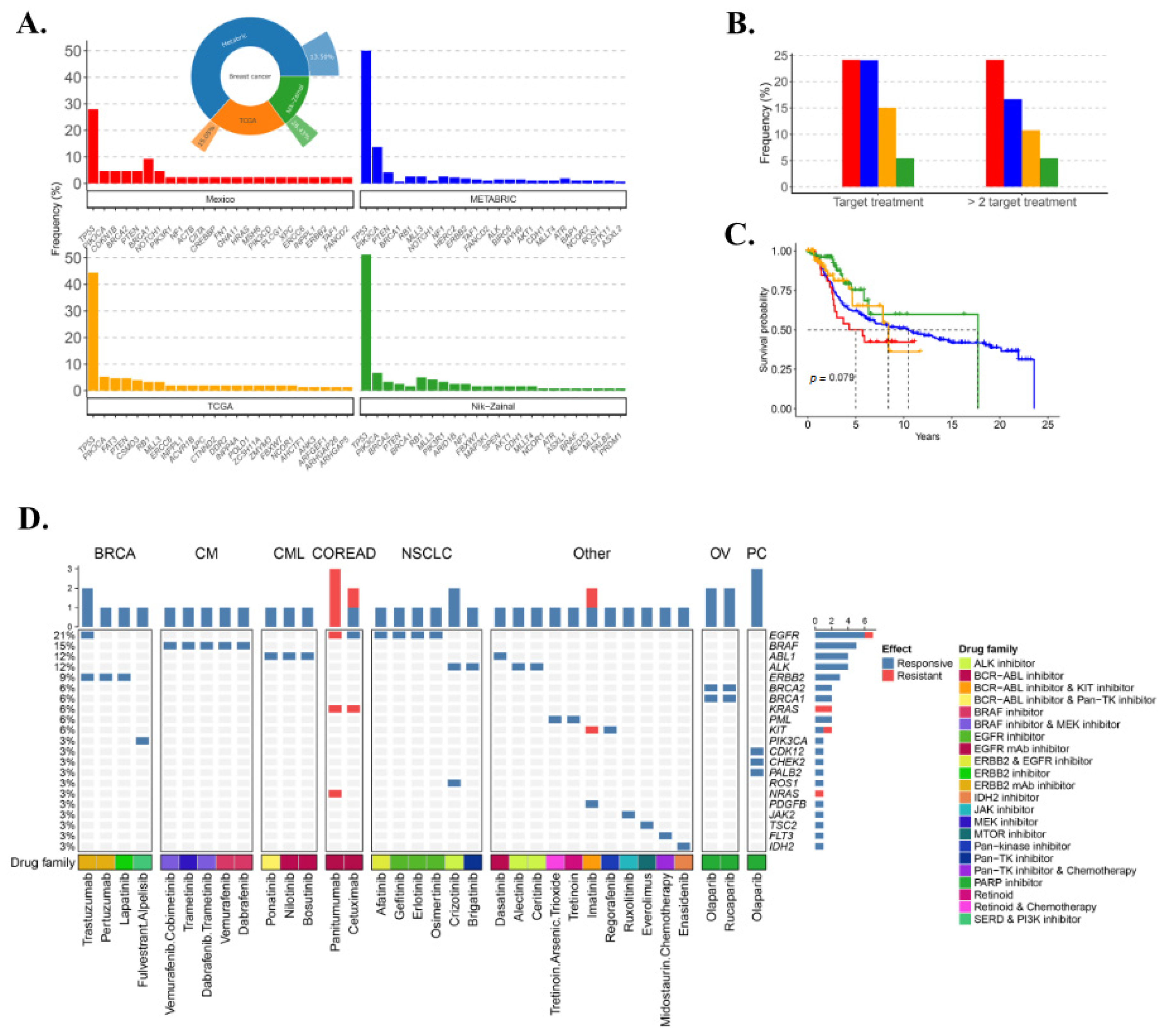
3.3. Mutational Burden
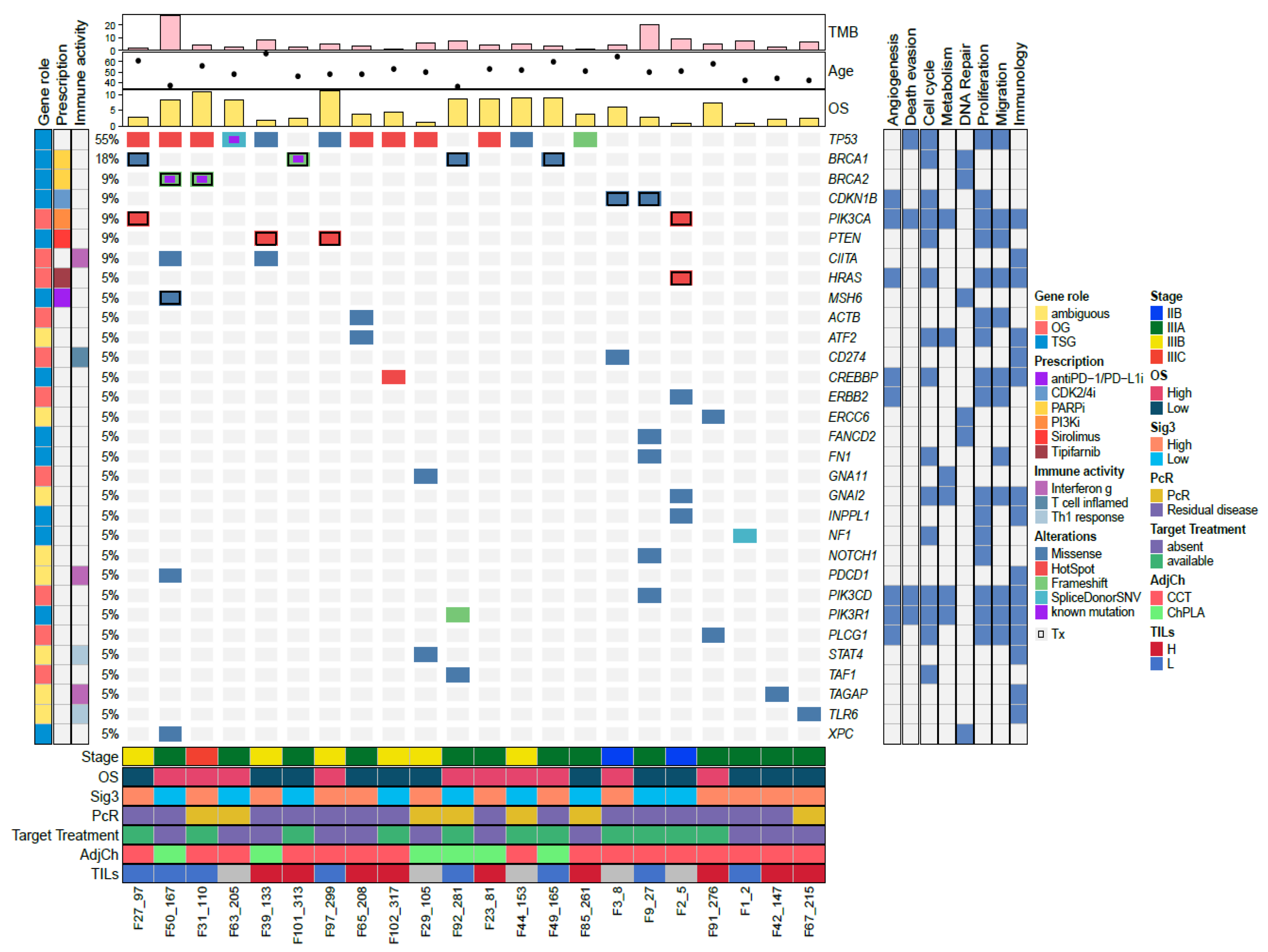
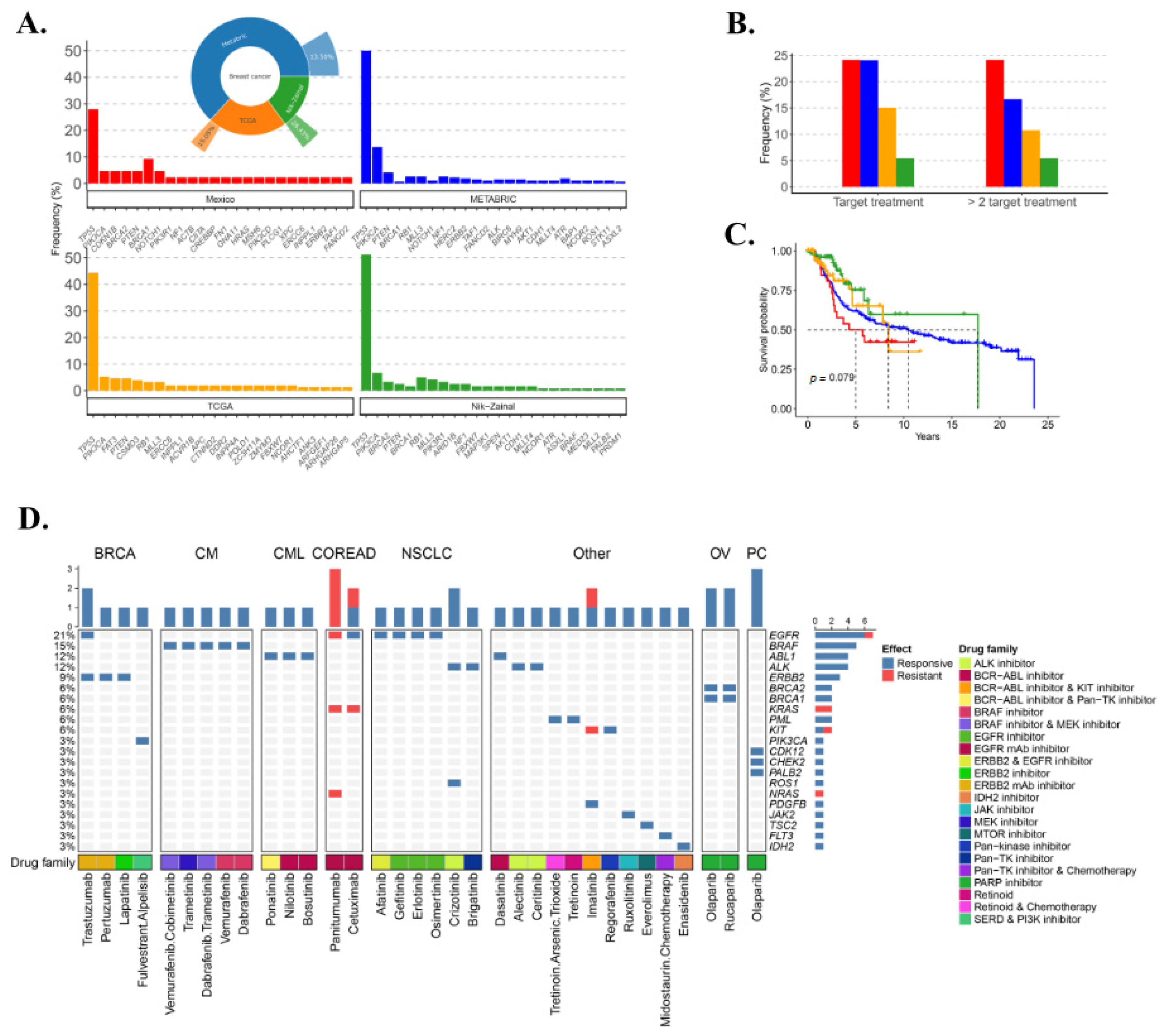
3.4. Somatic Mutational Landscape
3.5. Tumor Infiltrating Lymphocytes
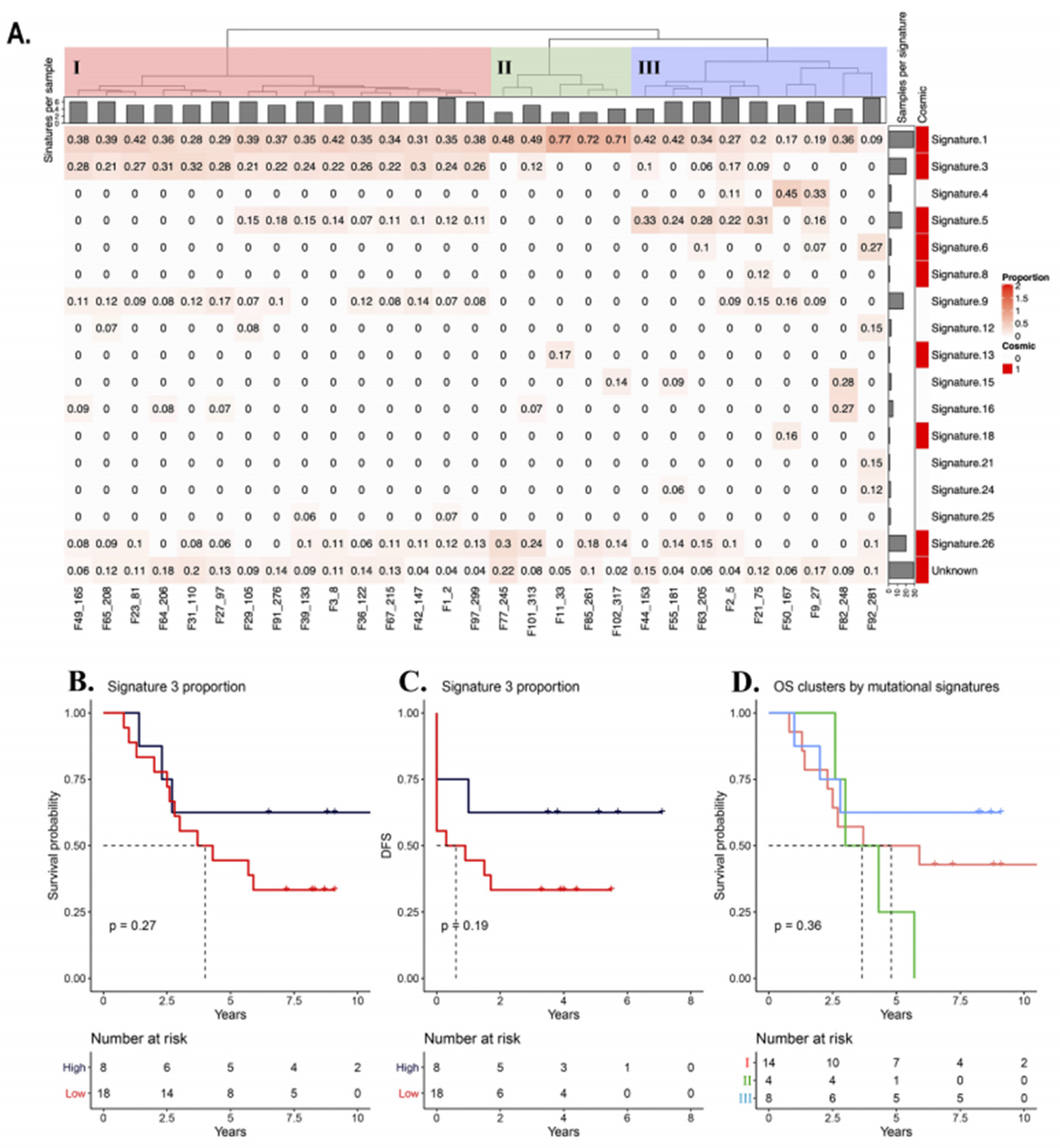
3.6. Mutational Signatures
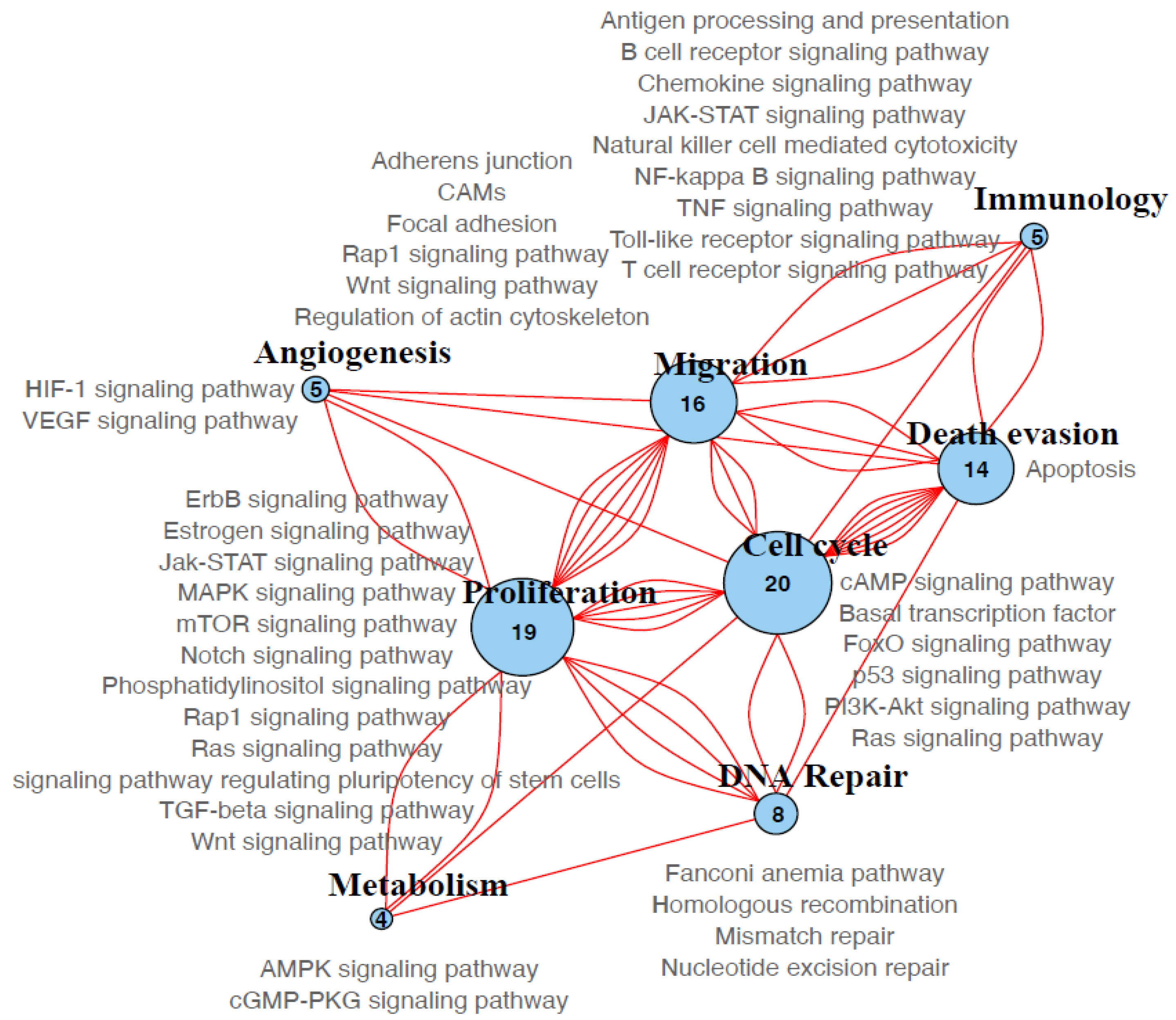
3.7. Molecular Pathways
3.8. TNBC Potential Therapy Perspective
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradishar, W.J.; Abraham, J.; Aft, R.; Agnese, D.; Allison, K.H.; Blair, S.L.; Burstein, H.J.; Dang, C.; Elias, A.D.; Giordano, S.H.; et al. NCCN guidelines insights: Breast cancer, version 3.2018. J. Natl. Compr. Cancer Netw. 2019, 17, 118–126. [Google Scholar]
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Minckwitz, G.; Schneeweiss, A.; Loibl, S.; Salat, C.; Denkert, C.; Rezai, M.; Blohmer, J.U.; Jackisch, C.; Paepke, S.; Gerber, B.; et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): A randomised phase 2 trial. Lancet Oncol. 2014, 15, 747–756. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef]
- Kwan, M.L.; Kushi, L.H.; Weltzien, E.; Maring, B.; Kutner, S.E.; Fulton, R.S.; Lee, M.M.; Ambrosone, C.B.; Caan, B.J. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009, 11, R31. [Google Scholar] [CrossRef]
- Lara-Medina, F.; Pérez-Sánchez, V.; Saavedra-Pérez, D.; Blake-Cerda, M.; Arce, C.; Motola-Kuba, D.; Villarreal-Garza, C.; González-Angulo, A.M.; Bargalló, E.; Aguilar, J.L.; et al. Triple-negative breast cancer in Hispanic patients. Cancer 2011, 117, 3658–3669. [Google Scholar] [CrossRef]
- Soto-Perez-de-Celis, E.; Chavarri-Guerra, Y. National and regional breast cancer incidence and mortality trends in Mexico 2001–2011: Analysis of a population-based database. Cancer Epidemiol. 2016, 41, 24–33. [Google Scholar] [CrossRef]
- Weitzel, J.N.; Neuhausen, S.L.; Adamson, A.; Tao, S.; Ricker, C.; Maoz, A.; Rosenblatt, M.; Nehoray, B.; Sand, S.; Steele, L.; et al. Pathogenic and likely pathogenic variants in PALB2, CHEK2, and other known breast cancer susceptibility genes among 1054 BRCA-negative Hispanics with breast cancer. Cancer 2019, 125, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Rueda, O.M.; Aparicio, S.; Caldas, C. A new genome-driven integrated classification of breast cancer and its implications. EMBO J. 2013, 32, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jézéquel, P.; Kerdraon, O.; Hondermarck, H.; Guérin-Charbonnel, C.; Lasla, H.; Gouraud, W.; Canon, J.-L.; Gombos, A.; Dalenc, F.; Delaloge, S.; et al. Identification of three subtypes of triple-negative breast cancer with potential therapeutic implications. Breast Cancer Res. 2019, 21, 65. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Brown, J.; Dent, R.; Jackisch, C.; Mackey, J.; Pivot, X.; Steger, G.G.; Suter, T.M.; Toi, M.; Parmar, M.; et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): Primary results of a randomised, phase 3 trial. Lancet Oncol. 2013, 14, 933–942. [Google Scholar] [CrossRef]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J Clin Oncol. 2020. [Google Scholar] [CrossRef]
- Telli, M.L.; Stover, D.G.; Loi, S.; Aparicio, S.; Carey, L.A.; Domchek, S.M.; Newman, L.; Sledge, G.W.; Winer, E.P.; Stover, D.G.; et al. Homologous recombination deficiency and host anti-tumor immunity in triple-negative breast cancer. Breast Cancer Res. Treat. 2018, 171, 21–31. [Google Scholar] [CrossRef]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.A.; Schmidt, M.K.; van Beers, E.H.; Cornelissen, S.; Holtkamp, M.; Froklage, F.E.; de Vries, E.G.E.; et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann. Oncol. 2011, 22, 1561–1570. [Google Scholar] [CrossRef]
- Barragán Ruíz, J.A.; Becerra Alcántara, G.I.; González López, N.J.; Mainero Ratchelous, F.E.; Martínez Mijares, A.; Patlán Pérez, R.M.; Pérez Puente, A.; Silva Juan, A. Tratamiento del cáncer mama. In Guía de Práctica Clínica Diagnóstico y Tratamiento del Cáncer de Mama en Segundo y Tercer Nivel de Atención; CENETEC: México City, México, 2009. [Google Scholar]
- Pathologic Complete Response in Neoadjuvant Treatment of High-Risk Early-Stage Breast Cancer: Use as an Endpoint to Support Accelerated Approval/FDA. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/pathologic-complete-response-neoadjuvant-treatment-high-risk-early-stage-breast-cancer-use-endpoint#search=’FDA+draft+guidance+neoadjuvant’ (accessed on 19 January 2020).
- Hammond, M.E.H.; Hayes, D.F.; Dowsett, M.; Allred, D.C.; Hagerty, K.L.; Badve, S.; Fitzgibbons, P.L.; Francis, G.; Goldstein, N.S.; Hayes, M.; et al. Frequently asked questions recommendations for HER2 testing in breast cancer: ASCO-CAP clinical practice guideline update background questions. Arch. Pathol. Lab. Med. 2010, 134, 907–922. [Google Scholar] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From fastQ data to high-confidence variant calls: The genome analysis toolkit best practices pipeline. In Current Protocols in Bioinformatics; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; Volume 43, pp. 11.10.1–11.10.33. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019, 531210. [Google Scholar] [CrossRef]
- Exome Variant Server. Available online: https://evs.gs.washington.edu/EVS/ (accessed on 28 January 2020).
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. Mutationtaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1084. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.; Kinzler, K.W.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Chtanova, T.; Tangye, S.G.; Newton, R.; Frank, N.; Hodge, M.R.; Rolph, M.S.; Mackay, C.R. T Follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J. Immunol. 2004, 173, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Grotz, T.E.; Jakub, J.W.; Mansfield, A.S.; Goldenstein, R.; Enninga, E.A.L.; Nevala, W.K.; Leontovich, A.A.; Markovic, S.N. Evidence of Th2 polarization of the sentinel lymph node (SLN) in melanoma. Oncoimmunology 2015, 4, e1026504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; et al. Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, R.; McGranahan, N.; Herrero, J.; Taylor, B.S.; Swanton, C. DeconstructSigs: Delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016, 17, 31. [Google Scholar] [CrossRef] [Green Version]
- Ward, J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- cBioPortal for Cancer Genomics. Available online: http://www.cbioportal.org/study?id=brca_tcga_pub2015&tab=summary (accessed on 30 January 2019).
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarty, D.; Gao, J.; Phillips, S.; Kundra, R.; Zhang, H.; Wang, J.; Rudolph, J.E.; Yaeger, R.; Soumerai, T.; Nissan, M.H.; et al. OncoKB: A precision oncology knowledge base. JCO Precis. Oncol. 2017, 1, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaca-Paniagua, F.; Alvarez-Gomez, R.M.; Maldonado-Martínez, H.A.; Pérez-Plasencia, C.; Fragoso-Ontiveros, V.; Lasa-Gonsebatt, F.; Herrera, L.A.; Cantú, D.; Bargallo-Rocha, E.; Mohar, A.; et al. Revealing the molecular portrait of triple negative breast tumors in an understudied population through Omics analysis of Formalin-fixed and Paraffin-embedded tissues. PLoS ONE 2015, 10, e0126762. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Reynoso-Noverón, N.; Villarreal-Garza, C.; Soto-Perez-de-Celis, E.; Arce-Salinas, C.; Matus-Santos, J.; Ramírez-Ugalde, M.T.; Alvarado-Miranda, A.; Cabrera-Galeana, P.; Meneses-García, A.; Lara-Medina, F.; et al. Clinical and epidemiological profile of breast cancer in Mexico: Results of the Seguro Popular. J. Glob. Oncol. 2017, 3, 757–764. [Google Scholar] [CrossRef]
- Flores-Díaz, D.; Arce, C.; Flores-Luna, L.; Reynoso-Noveron, N.; Lara-Medina, F.; Matus, J.A.; Bargallo-Rocha, E.; Pérez, V.; Villarreal-Garza, C.; Cabrera-Galeana, P.; et al. Impact of invasive lobular carcinoma on long-term outcomes in Mexican breast cancer patients. Breast Cancer Res. Treat. 2019, 176, 243–249. [Google Scholar] [CrossRef]
- Parise, C.A.; Caggiano, V. Breast cancer survival defined by the er/pr/her2 subtypes and a surrogate classification according to tumor grade and immunohistochemical biomarkers. J. Cancer Epidemiol. 2014, 2014. [Google Scholar] [CrossRef]
- Spitale, A.; Mazzola, P.; Soldini, D.; Mazzucchelli, L.; Bordoni, A. Breast cancer classification according to immunohistochemical markers: Clinicopathologic features and short-term survival analysis in a population-based study from the South of Switzerland. Ann. Oncol. 2009, 20, 628–635. [Google Scholar] [CrossRef]
- Alba, E.; Chacon, J.I.; Lluch, A.; Anton, A.; Estevez, L.; Cirauqui, B.; Carrasco, E.; Calvo, L.; Segui, M.A.; Ribelles, N.; et al. A randomized phase II trial of platinum salts in basal-like breast cancer patients in the neoadjuvant setting. Results from the GEICAM/2006-03, multicenter study. Breast Cancer Res. Treat. 2012, 136, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Glaspy, J.; Allison, M.A.; Kass, F.C.; Elashoff, R.; Chung, D.U.; Gornbein, J. Differential response of triple-negative breast cancer to a docetaxel and carboplatin-based neoadjuvant treatment. Cancer 2010, 116, 4227–4237. [Google Scholar] [CrossRef] [PubMed]
- Roy, V.; Pockaj, B.A.; Allred, J.B.; Apsey, H.; Northfelt, D.W.; Nikcevich, D.; Mattar, B.; Perez, E.A. A phase II trial of docetaxel and carboplatin administered every 2 weeks as preoperative therapy for stage II or III breast cancer: NCCTG study N0338. Am. J. Clin. Oncol. Cancer Clin. Trials 2013, 36, 540–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikov, W.M.; Berry, D.A.; Perou, C.M.; Singh, B.; Cirrincione, C.T.; Tolaney, S.M.; Kuzma, C.S.; Pluard, T.J.; Somlo, G.; Port, E.R.; et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J. Clin. Oncol. 2015, 33, 13–21. [Google Scholar]
- Silver, D.P.; Richardson, A.L.; Eklund, A.C.; Wang, Z.C.; Szallasi, Z.; Li, Q.; Juul, N.; Leong, C.-O.; Calogrias, D.; Buraimoh, A.; et al. Efficacy of neoadjuvant cisplatin in triple-negative breast cancer. J. Clin. Oncol. 2010, 28, 1145–1153. [Google Scholar] [CrossRef]
- Zhang, P.; Yin, Y.; Mo, H.; Zhang, B.; Wang, X.; Li, Q.; Yuan, P.; Wang, J.; Zheng, S.; Cai, R.; et al. Better pathologic complete response and relapse-free survival after carboplatin plus paclitaxel compared with epirubicin plus paclitaxel as neoadjuvant chemotherapy for locally advanced triple-negative breast cancer: A randomized phase 2 trial. Oncotarget 2016, 7, 60647–60656. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.M.; Kim, R.N.; Kwon, M.J.; Oh, E.; Han, J.; Lee, S.K.; Choi, J.S.; Park, S.; Nam, S.J.; Gong, G.Y.; et al. Targeted exome sequencing of Korean triple-negative breast cancer reveals homozygous deletions associated with poor prognosis of adjuvant chemotherapy-treated patients. Oncotarget 2017, 8, 61538–61550. [Google Scholar] [CrossRef] [Green Version]
- Martelotto, L.G.; De Filippo, M.R.; Ng, C.K.; Natrajan, R.; Fuhrmann, L.; Cyrta, J.; Piscuoglio, S.; Wen, H.C.; Lim, R.S.; Shen, R.; et al. Genomic landscape of adenoid cystic carcinoma of the breast. J. Pathol. 2015, 237, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. New Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legerski, R.; Peterson, C. Expression cloning of a human DNA repair gene involved in xeroderma pigmentosum group C. Nature 1992, 359, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell 2018, 173, 879–893.e13. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Rodler, E.T.; Kurland, B.F.; Griffin, M.; Gralow, J.R.; Porter, P.; Yeh, R.F.; Gadi, V.K.; Guenthoer, J.; Beumer, J.H.; Korde, L.; et al. Phase I Study of Veliparib (ABT-888) Combined with Cisplatin and Vinorelbine in advanced triple-negative breast cancer and/or BRCA M-mutation-associated breast cancer. Clin. Cancer Res. 2016, 22, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Janku, F.; Wheler, J.J.; Westin, S.N.; Moulder, S.L.; Naing, A.; Tsimberidou, A.M.; Fu, S.; Falchook, G.S.; Hong, D.S.; Garrido-Laguna, I.; et al. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J. Clin. Oncol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Juric, D.; Krop, I.; Ramanathan, R.K.; Wilson, T.R.; Ware, J.A.; Sanabria Bohorquez, S.M.; Savage, H.M.; Sampath, D.; Salphati, L.; Lin, R.S.; et al. Phase I dose-escalation study of taselisib, an oral PI3K inhibitor, in patients with advanced solid tumors. Cancer Discov. 2017, 7, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef]
- Stein, E.; Chromik, J.; DeAngelo, D.J.; Chatterjee, M.; Noppeney, R.; de Vos, F.; Minami, H.; Jeay, S.; Meille, C.; Halilovic, E.; et al. Abstract CT152: Phase I dose- and regimen-finding study of NVP-HDM201 in pts with advanced TP53 wt acute leukemias. In Proceedings of the Cancer Research, American Association for Cancer Research (AACR), Washington, DC, USA, 1–5 April 2017; Volume 77, p. CT152. [Google Scholar]
- Ricciardi, M.R.; Scerpa, M.C.; Bergamo, P.; Ciuffreda, L.; Petrucci, M.T.; Chiaretti, S.; Tavolaro, S.; Mascolo, M.G.; Abrams, S.L.; Steelman, L.S.; et al. Therapeutic potential of MEK inhibition in acute myelogenous leukemia: Rationale for “vertical” and “lateral” combination strategies. J. Mol. Med. 2012, 90, 1133–1144. [Google Scholar] [CrossRef]
- Daver, N.; Cortes, J. Molecular targeted therapy in acute myeloid leukemia. Hematology 2012, 17, s59–s62. [Google Scholar] [CrossRef]
- Yap, T.A.; Yan, L.; Patnaik, A.; Fearen, I.; Olmos, D.; Papadopoulos, K.; Baird, R.D.; Delgado, L.; Taylor, A.; Lupinacci, L.; et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-206 in patients with advanced solid tumors. J. Clin. Oncol. 2011, 29, 4688–4695. [Google Scholar] [CrossRef] [PubMed]
- Stone, A.; Sutherland, R.L.; Musgrove, E.A. Inhibitors of cell cycle kinases: Recent advances and future prospects as cancer therapeutics. Crit. Rev. Oncog. 2012, 17, 175–198. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, W.; Wali, V.B.; Pongor, L.S.; Li, C.; Lau, R.; Győrffy, B.; Lifton, R.P.; Symmans, W.F.; Pusztai, L.; et al. Predictors of chemosensitivity in triple negative breast cancer: An integrated genomic analysis. PLoS Med. 2016, 13, e1002193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.C.; Novik, Y.; Stein, S.; Volm, M.; Meyers, M.; Smith, J.; Omene, C.; Speyer, J.; Schneider, R.; Jhaveri, K.; et al. Phase 2 trial of everolimus and carboplatin combination in patients with triple negative metastatic breast cancer. Breast Cancer Res. 2014, 16, R32. [Google Scholar] [CrossRef] [Green Version]
- Staaf, J.; Glodzik, D.; Bosch, A.; Vallon-Christersson, J.; Reuterswärd, C.; Häkkinen, J.; Degasperi, A.; Amarante, T.D.; Saal, L.H.; Hegardt, C.; et al. Whole-genome sequencing of triple-negative breast cancers in a population-based clinical study. Nat. Med. 2019, 25, 1526–1533. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and transcriptomic landscape of triple-negative breast cancers: Subtypes and treatment strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [Green Version]
- Adams, S.; Gray, R.J.; Demaria, S.; Goldstein, L.; Perez, E.A.; Shulman, L.N.; Martino, S.; Wang, M.; Jones, V.E.; Saphner, T.J.; et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J. Clin. Oncol. 2014, 32, 2959–2966. [Google Scholar] [CrossRef]
- Hutchinson, K.E.; Yost, S.E.; Chang, C.-W.; Johnson, R.M.; Carr, A.R.; McAdam, P.R.; Halligan, D.L.; Chang, C.-C.; Schmolze, D.; Liang, J.; et al. Comprehensive profiling of poor-risk paired primary and recurrent triple-negative breast cancers reveals immune phenotype shifts. Clin. Cancer Res. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Gardner, S.A.; Hovhannisyan, H.; Natalizio, A.; Weymouth, K.S.; Chen, W.; Thibodeau, I.; Bogdanova, E.; Letovsky, S.; Willis, A.; et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: Insights of relevance to variant classification. Genet. Med. 2016, 18, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, Y.; Kanai, M.; Kou, T.; Sugiyama, A.; Nakamura, E.; Miyake, H.; Yamada, T.; Nishigaki, M.; Kondo, T.; Murakami, H.; et al. Clinical significance of TP53 variants as possible secondary findings in tumor-only next-generation sequencing. J. Hum. Genet. 2019, 65, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Birgisdottir, V.; Stefansson, O.A.; Bodvarsdottir, S.K.; Hilmarsdottir, H.; Jonasson, J.G.; Eyfjord, J.E. Epigenetic silencing and deletion of the BRCA1 gene in sporadic breast cancer. Breast Cancer Res. 2006, 8, R38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldeira, J.R.F.; Prando, É.C.; Quevedo, F.C.; Moraes Neto, F.A.; Rainho, C.A.; Rogatto, S.R. CDH1 promoter hypermethylation and E-cadherin protein expression in infiltrating breast cancer. BMC Cancer 2006, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.; Tamura, G.; Tsuchiya, T.; Sakata, K.; Kashiwaba, M.; Osakabe, M.; Motoyama, T. Adenomatous polyposis coli (APC) gene promoter hypermethylation in primary breast cancers. Br. J. Cancer 2001, 85, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Wei, M.; Xu, J.; Dignam, J.; Nanda, R.; Sveen, L.; Fackenthal, J.; Grushko, T.A.; Olopade, O.I. Estrogen receptor α, BRCA1, and FANCF promoter methylation occur in distinct subsets of sporadic breast cancers. Breast Cancer Res. Treat. 2008, 111, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Kawazu, M.; Kojima, S.; Ueno, T.; Totoki, Y.; Nakamura, H.; Kunita, A.; Qu, W.; Yoshimura, J.; Soda, M.; Yasuda, T.; et al. Integrative analysis of genomic alterations in triple-negative breast cancer in association with homologous recombination deficiency. PLoS Genet. 2017, 13. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Lee, J.S.; Yost, S.E.; Blanchard, S.; Schmolze, D.; Yin, H.H.; Pillai, R.; Robinson, K.; Tang, A.; Martinez, N.; Portnow, J.; et al. Phase I clinical trial of the combination of eribulin and everolimus in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2019, 21, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Deng, H.; Rao, N.; You, N.; Yang, Y.; Cao, M.; Liu, J. Neoadjuvant everolimus plus letrozole versus fluorouracil, epirubicin and cyclophosphamide for ER-positive, HER2-negative breast cancer: Study protocol for a randomized pilot trial. Trials 2017, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanovic, B.; Mayer, I.A.; Mayer, E.L.; Abramson, V.G.; Bardia, A.; Sanders, M.E.; Kuba, M.G.; Estrada, M.V.; Beeler, J.S.; Shaver, T.M.; et al. A randomized phase II neoadjuvant study of cisplatin, paclitaxel with or without everolimus in patients with stage II/III triple-negative breast cancer (TNBC): Responses and long-term outcome correlated with increased frequency of DNA damage response gene mutations, TNBC subtype, AR status, and Ki67. Clin. Cancer Res. 2017, 23, 4035–4045. [Google Scholar] [PubMed] [Green Version]
- Roviello, G.; Milani, M.; Gobbi, A.; Cappelletti, M.R.; Zanotti, L.; Senti, C.; Bottini, A.; Strina, C.; Sigala, S.; Generali, D. A Phase Ib Open-Label Study to Assess the Safety and Tolerability of Everolimus in Combination with Eribulin in Triple-Negative Breast Cancers. Clin. Breast Cancer 2016, 16, e57–e59. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Akcakanat, A.; Liu, S.; Green, M.C.; Murray, J.L.; Chen, H.; Palla, S.L.; Koenig, K.B.; Brewster, A.M.; Valero, V.; et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancer. Ann. Oncol. 2014, 25, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Bernsdorf, M.; Ingvar, C.; Jörgensen, L.; Tuxen, M.K.; Jakobsen, E.H.; Saetersdal, A.; Kimper-Karl, M.L.; Kroman, N.; Balslev, E.; Ejlertsen, B. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res. Treat. 2011, 126, 463–470. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Hannemann, J.; Laddach, N.; Vrancken Peeters, M.T.F.D.; van de Vijver, M.J.; Wesseling, J.; Nederlof, P.M.; Rodenhuis, S. Indicators of homologous recombination deficiency in breast cancer and association with response to neoadjuvant chemotherapy. Ann. Oncol. 2011, 22, 870–876. [Google Scholar] [CrossRef]
- Lips, E.H.; Mulder, L.; Oonk, A.; Van Der Kolk, L.E.; Hogervorst, F.B.L.; Imholz, A.L.T.; Wesseling, J.; Rodenhuis, S.; Nederlof, P.M. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br. J. Cancer 2013, 108, 2172–2177. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age at the Diagnosis | Hereditary Breast Cancer | ||
|---|---|---|---|
| Mean | 50.1 (± 8.72) | Yes | 3 (10.34%) |
| Range | 29–68 | No | 26 (89.66%) |
| Menopausal status | Tumor stage at diagnosis | ||
| Premenopausal | 10 (34%) | IIB | 5 (17.24%) |
| Postmenopausal | 16 (55%) | IIIA | 14 (48.28%) |
| Co-morbidities | IIIB | 7 (24.24%) | |
| Diabetes mellitus | 7 (24%) | IIIC | 3 (10.34%) |
| Systemic hypertension | 7 (24%) | Pathological complete response | |
| BMC * | PCR | 10 (34.48%) | |
| Mean | 27.94 (± 5.10) | Residual Disease | 19 (65.52%) |
| Previous pregnancy | Neoadjuvant chemotherapy with platinum salts | ||
| Yes | 24 (82.76%) | Yes | 10 (34.48%) |
| No | 5 (17.24%) | No | 19 (65.51%) |
| Oral contraceptives | DFS * (years) | ||
| Yes | 7 (24.24%) | Mean | 2 (± 2.24) |
| No | 22 (75.86%) | Range | 0–7.1 |
| OS * (years) | |||
| Mean | 5 (± 2.24) | ||
| Range | 0.7–11.1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas-Jiménez, E.; Mejía-Gómez, J.C.; Díaz-Velásquez, C.; Quezada-Urban, R.; Martínez Gregorio, H.; Vallejo-Lecuona, F.; de la Cruz-Montoya, A.; Porras Reyes, F.I.; Pérez-Sánchez, V.M.; Maldonado-Martínez, H.A.; et al. Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic. Genes 2020, 11, 1367. https://doi.org/10.3390/genes11111367
Rojas-Jiménez E, Mejía-Gómez JC, Díaz-Velásquez C, Quezada-Urban R, Martínez Gregorio H, Vallejo-Lecuona F, de la Cruz-Montoya A, Porras Reyes FI, Pérez-Sánchez VM, Maldonado-Martínez HA, et al. Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic. Genes. 2020; 11(11):1367. https://doi.org/10.3390/genes11111367
Chicago/Turabian StyleRojas-Jiménez, Ernesto, Javier César Mejía-Gómez, Clara Díaz-Velásquez, Rosalía Quezada-Urban, Héctor Martínez Gregorio, Fernando Vallejo-Lecuona, Aldo de la Cruz-Montoya, Fany Iris Porras Reyes, Víctor Manuel Pérez-Sánchez, Héctor Aquiles Maldonado-Martínez, and et al. 2020. "Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic" Genes 11, no. 11: 1367. https://doi.org/10.3390/genes11111367
APA StyleRojas-Jiménez, E., Mejía-Gómez, J. C., Díaz-Velásquez, C., Quezada-Urban, R., Martínez Gregorio, H., Vallejo-Lecuona, F., de la Cruz-Montoya, A., Porras Reyes, F. I., Pérez-Sánchez, V. M., Maldonado-Martínez, H. A., Robles-Estrada, M., Bargalló-Rocha, E., Cabrera-Galeana, P., Ramos-Ramírez, M., Chirino, Y. I., Alonso Herrera, L., Terrazas, L. I., Oliver, J., Frecha, C., ... Vaca-Paniagua, F. (2020). Comprehensive Genomic Profile of Heterogeneous Long Follow-Up Triple-Negative Breast Cancer and Its Clinical Characteristics Shows DNA Repair Deficiency Has Better Prognostic. Genes, 11(11), 1367. https://doi.org/10.3390/genes11111367