PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction, Real-Time Quantitative PCR (qRT-PCR) and RNA Sequencing (RNA-seq)
2.3. ChIP-seq and ChIP-PCR Assay
2.4. Western Blot Analysis
2.5. Plasmid Construction
2.6. RNAi Interference
2.7. Transfection and Luciferase Assay
2.8. Metabolic Phenotypes
2.9. Statistical Analysis
3. Results
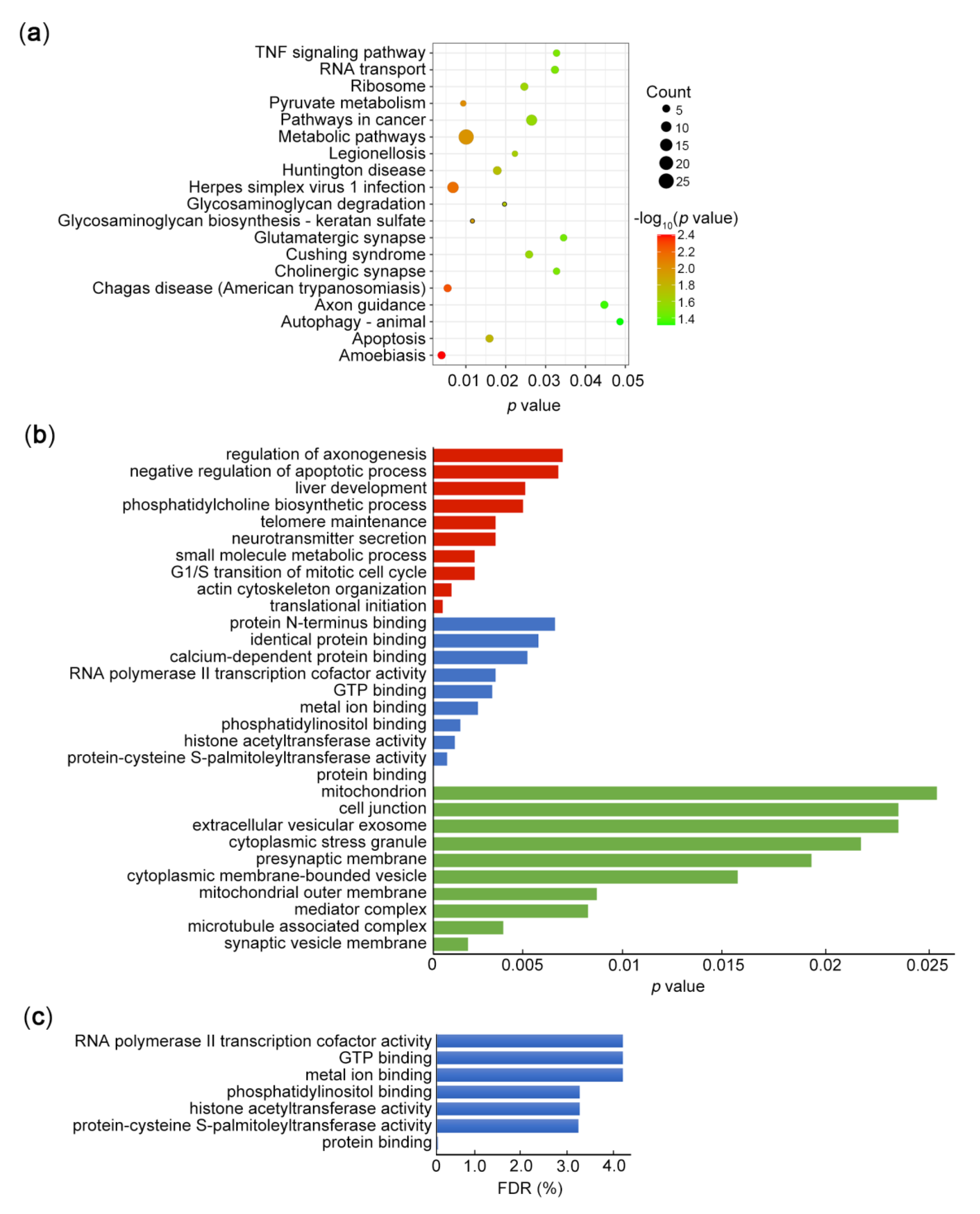
3.1. Motif and Function Analysis of ChIP-seq
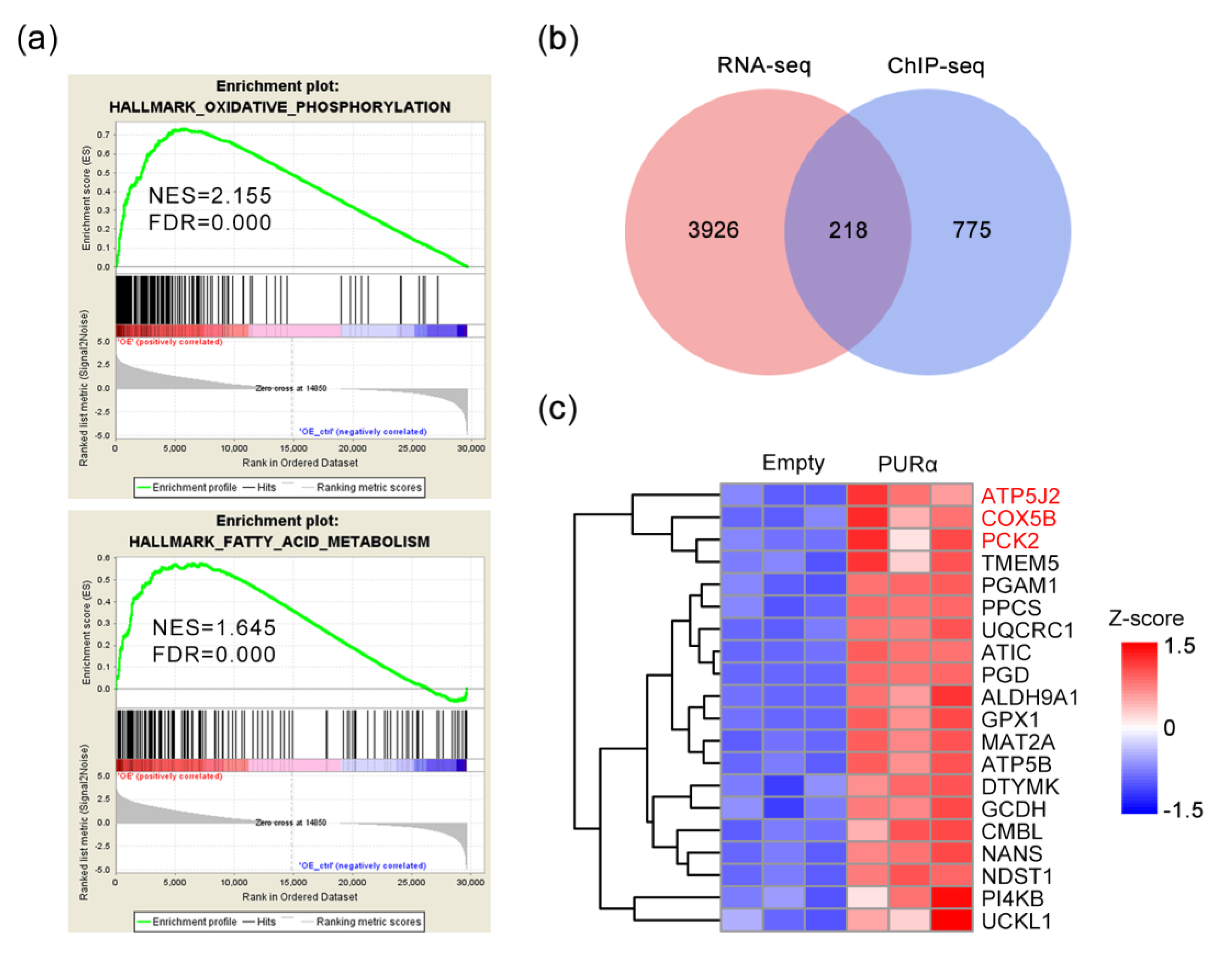
3.2. Screening of Potential Candidate Regulated Genes for PURα
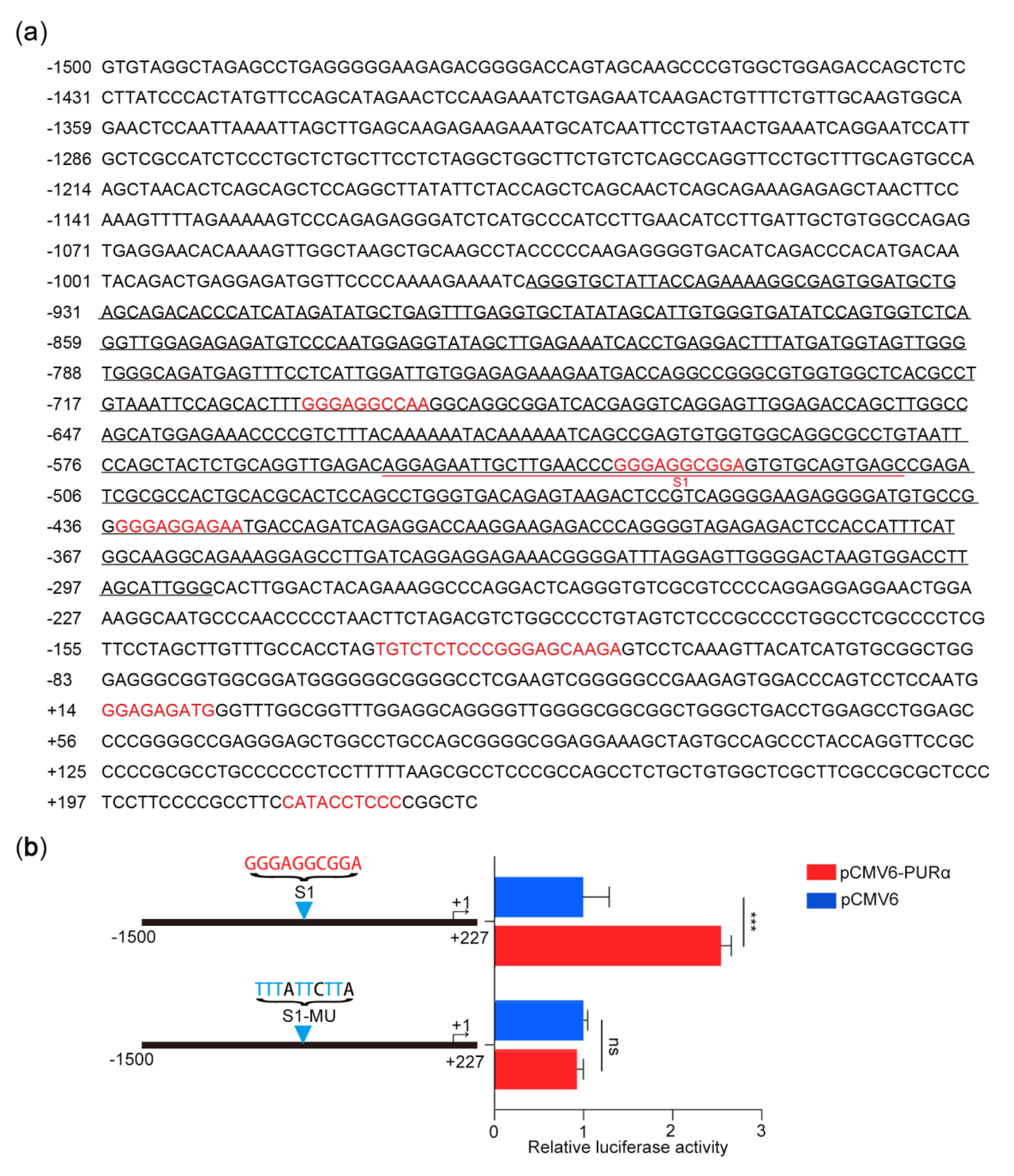
3.3. Transcriptional Activation of thePCK2 Promoter Depends on the GGN Motif bound by PURα
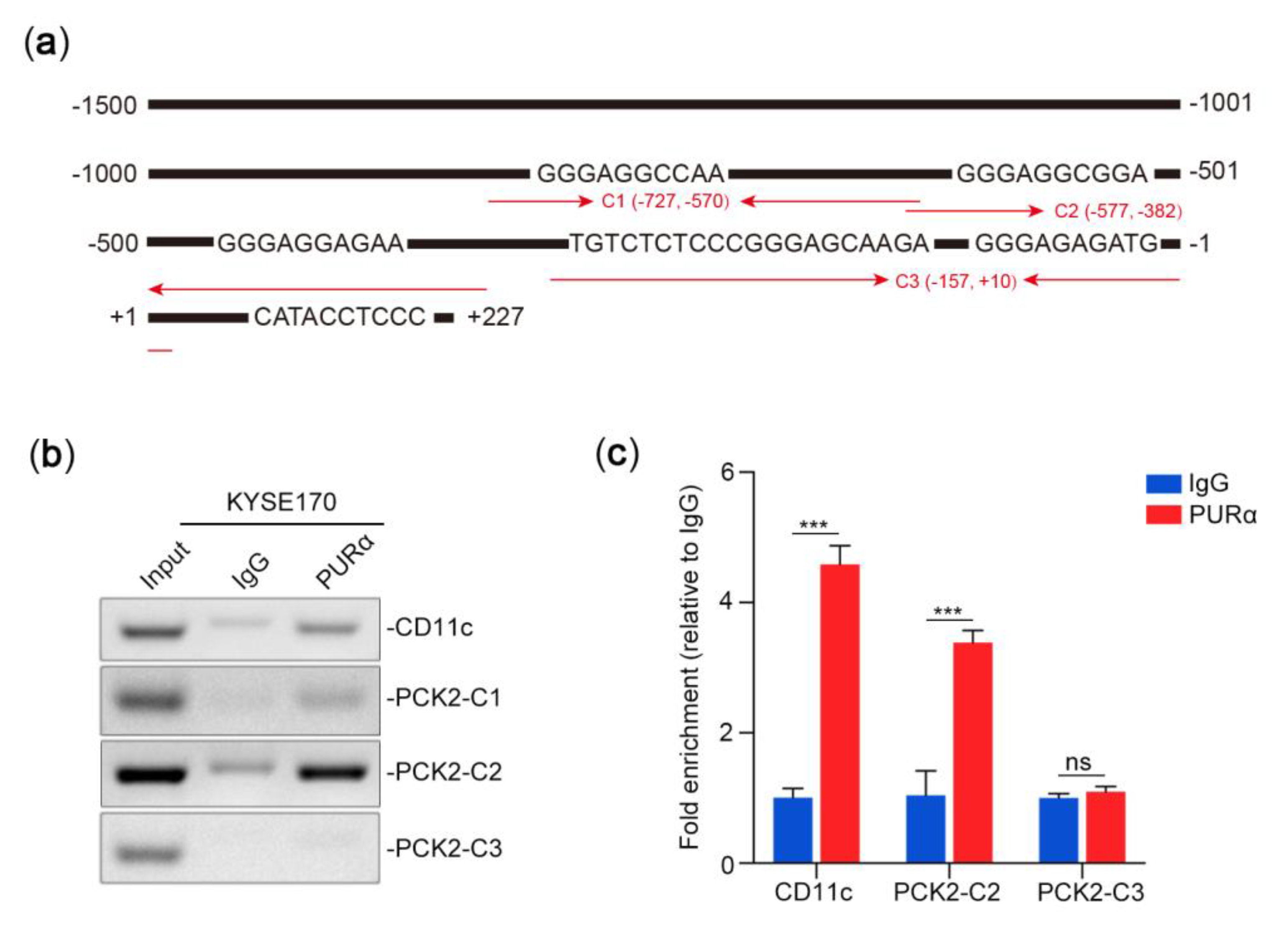
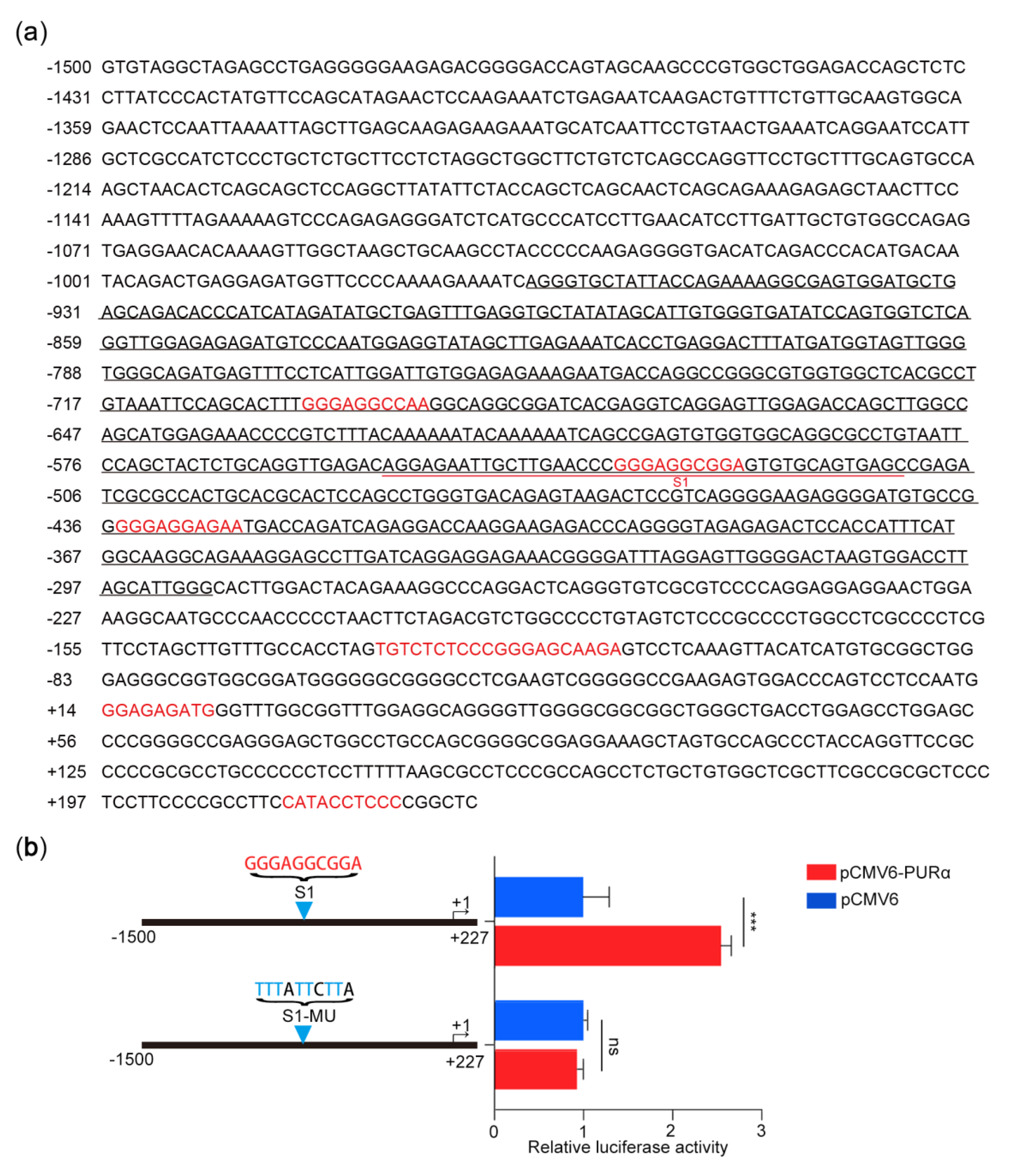
3.4. Identification of the Core Binding Region of the PCK2 Gene for PURα
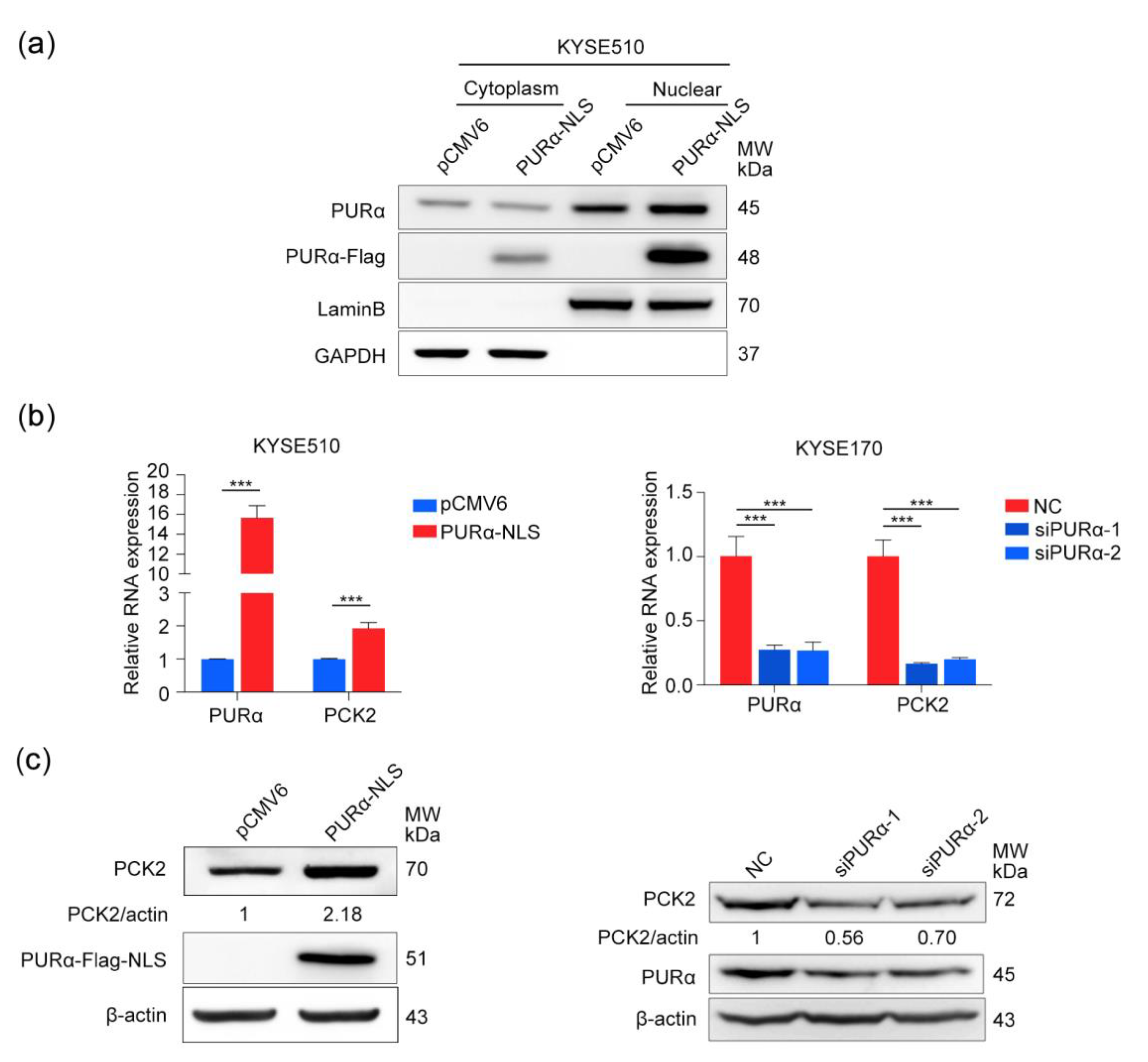
3.5. PURα Activates the Transcription and Expression of PCK2
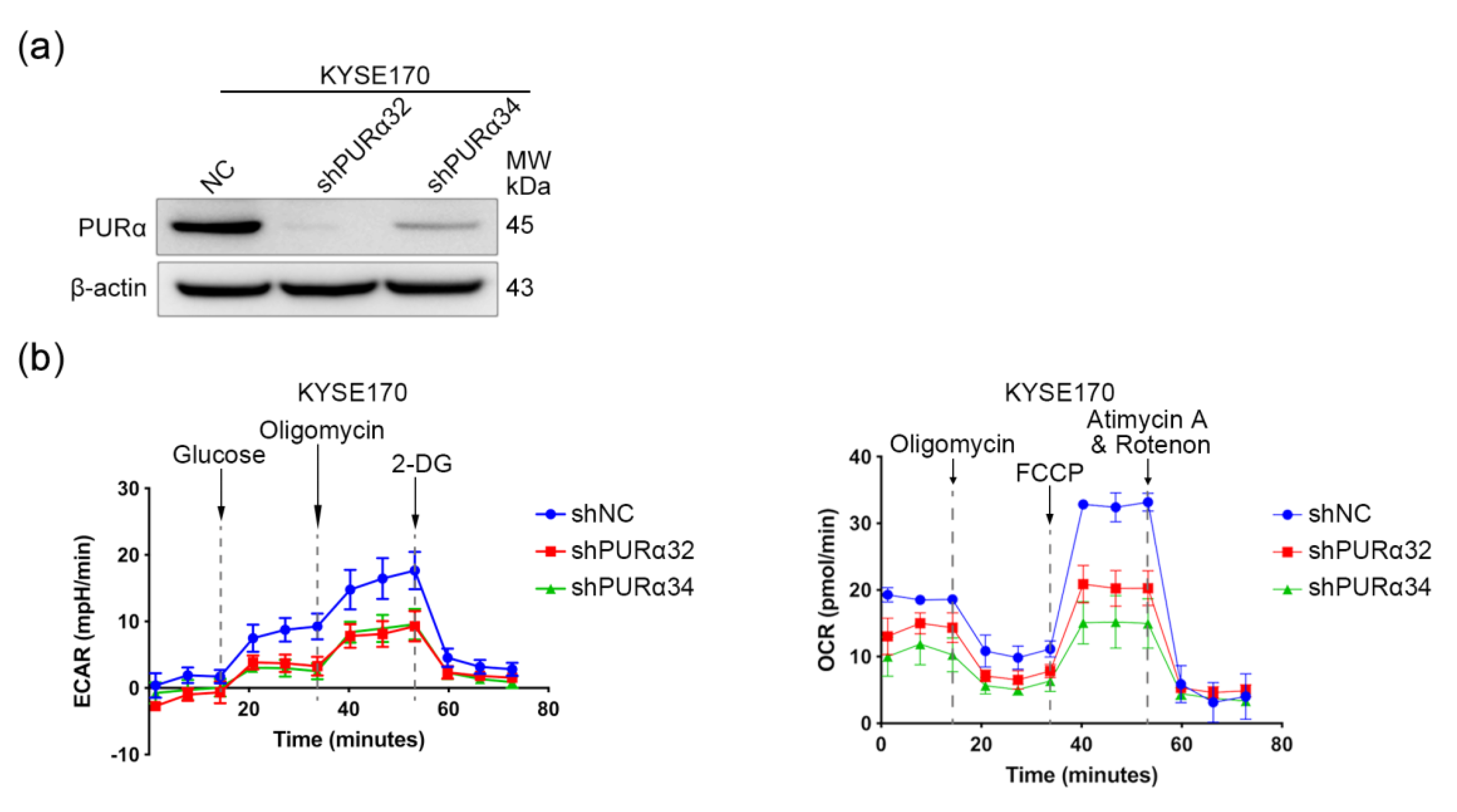
3.6. PURα Promotes the Mitochondrial Respiration and Glycolysis in ECSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zeng, H.; Chen, W.; Zheng, R.; Zhang, S.; Ji, J.S.; Zou, X.; Xia, C.; Sun, K.; Yang, Z.; Li, H.; et al. Changing cancer survival in China during 2003–15: A pooled analysis of 17 population-based cancer registries. Lancet Glob. Health 2018, 6, e555–e567. [Google Scholar] [CrossRef] [Green Version]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.L.; Santoni, G.; Ness-Jensen, E.; Lagergren, J.; Xie, S.H. Association Between Metformin Use and Risk of Esophageal Squamous Cell Carcinoma in a Population-Based Cohort Study. Am. J. Gastroenterol. 2020, 115, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Deng, M.; Xue, N.N.; Li, T.X.; Guo, Y.X.; Gao, L.; Zhao, D.; Fan, R.T. lncRNA KLF3-AS1 Suppresses Cell Migration and Invasion in ESCC by Impairing miR-185-5p-Targeted KLF3 Inhibition. Mol. Nucleic Acids 2020, 20, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Xu, E.; Bi, Y.; Xu, X.; Liu, X.; Song, B.; Zhang, L.; Cheng, C.; Yan, T.; Qian, Y.; et al. Novel ESCC-related gene ZNF750 as potential Prognostic biomarker and inhibits Epithelial-Mesenchymal Transition through directly depressing SNAI1 promoter in ESCC. Theranostics 2020, 10, 1798–1813. [Google Scholar] [CrossRef] [PubMed]
- Daniel, D.C.; Johnson, E.M. PURA, the gene encoding Pur-alpha, member of an ancient nucleic acid-binding protein family with mammalian neurological functions. Gene 2018, 643, 133–143. [Google Scholar] [CrossRef]
- Pandey, P.R.; Yang, J.H.; Tsitsipatis, D.; Panda, A.C.; Noh, J.H.; Kim, K.M.; Munk, R.; Nicholson, T.; Hanniford, D.; Argibay, D.; et al. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res. 2020, 48, 3789–3805. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.-M.; Li, W.; Zhao, X.-H. Purine-rich element binding protein alpha promotes invasion and migration of esophageal squamous cell carcinoma KYSE 510 cells by inducing epithelial-mesenchymal transition. World Chin. J. Dig. 2014, 22. [Google Scholar] [CrossRef]
- Kaminski, R.; Darbinyan, A.; Merabova, N.; Deshmane, S.L.; White, M.K.; Khalili, K. Protective role of Puralpha to cisplatin. Cancer Biol. 2008, 7, 1926–1935. [Google Scholar] [CrossRef] [Green Version]
- Bergemann, A.D.; Ma, Z.W.; Johnson, E.M. Sequence of cDNA comprising the human pur gene and sequence-specific single-stranded-DNA-binding properties of the encoded protein. Mol. Cell. Biol. 1992, 12, 5673–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Maeno, A.; Talbot, C., Jr.; Zeng, Y.; Yeater, D.B.; Leman, E.S.; Kulkarni, P.; Ogawa, O.; Getzenberg, R.H. Purine-rich element binding protein (PUR) alpha induces endoplasmic reticulum stress response, and cell differentiation pathways in prostate cancer cells. Prostate 2009, 69, 861–873. [Google Scholar] [CrossRef]
- Sariyer, I.K.; Sariyer, R.; Otte, J.; Gordon, J. Pur-Alpha Induces JCV Gene Expression and Viral Replication by Suppressing SRSF1 in Glial Cells. PLoS ONE 2016, 11, e0156819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Z.W.; Bergemann, A.D.; Johnson, E.M. Conservation in human and mouse Purα of a motif common to several proteins involved in initiation of DNA replication. Gene 1994, 149, 311–314. [Google Scholar] [CrossRef]
- Hokkanen, S.; Feldmann, H.M.; Ding, H.; Jung, C.K.; Bojarski, L.; Renner-Muller, I.; Schuller, U.; Kretzschmar, H.; Wolf, E.; Herms, J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 2012, 21, 473–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, K.; Del Valle, L.; Muralidharan, V.; Gault, W.J.; Darbinian, N.; Otte, J.; Meier, E.; Johnson, E.M.; Daniel, D.C.; Kinoshita, Y.; et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell. Biol. 2003, 23, 6857–6875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trau, S.P.; Pizoli, C.E. PURA Syndrome and Myotonia. Pediatr. Neurol. 2020, 104, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Reijnders, M.R.F.; Janowski, R.; Alvi, M.; Self, J.E.; van Essen, T.J.; Vreeburg, M.; Rouhl, R.P.W.; Stevens, S.J.C.; Stegmann, A.P.A.; Schieving, J.; et al. PURA syndrome: Clinical delineation and genotype-phenotype study in 32 individuals with review of published literature. J. Med. Genet. 2018, 55, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.H.; Reijnders, M.R.F.; Abubakare, O.; Tuttle, E.; Lape, B.; Minks, K.Q.; Stodgell, C.; Bennetto, L.; Kwon, J.; Fong, C.T.; et al. Expanding the neurodevelopmental phenotype of PURA syndrome. Am. J. Med. Genet. A 2018, 176, 56–67. [Google Scholar] [CrossRef]
- Lezon-Geyda, K.; Najfeld, V.; Johnson, E.M. Deletions of PURA, at 5q31, and PURB, at 7p13, in myelodysplastic syndrome and progression to acute myelogenous leukemia. Leukemia 2001, 15, 954–962. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Gomez-Pinillos, A.; Liu, X.; Johnson, E.M.; Ferrari, A.C. Induction of bicalutamide sensitivity in prostate cancer cells by an epigenetic Puralpha-mediated decrease in androgen receptor levels. Prostate 2010, 70, 179–189. [Google Scholar] [CrossRef]
- Urzua, U.; Roby, K.F.; Gangi, L.M.; Cherry, J.M.; Powell, J.I.; Munroe, D.J. Transcriptomic analysis of an in vitro murine model of ovarian carcinoma: Functional similarity to the human disease and identification of prospective tumoral markers and targets. J. Cell Physiol. 2006, 206, 594–602. [Google Scholar] [CrossRef]
- Wang, M.; An, S.; Wang, D.; Ji, H.; Geng, M.; Guo, X.; Wang, Z. Quantitative Proteomics Identify the Possible Tumor Suppressive Role of Protease-Activated Receptor-4 in Esophageal Squamous Cell Carcinoma Cells. Pathol. Oncol. Res. 2019, 25, 937–943. [Google Scholar] [CrossRef]
- Sharma, P.; Saraya, A.; Sharma, R. Potential diagnostic implications of miR-144 overexpression in human oesophageal cancer. Indian J. Med. Res. 2016, 143, S91–S103. [Google Scholar] [CrossRef]
- Walker, R.C.; Underwood, T.J. Molecular pathways in the development and treatment of oesophageal cancer. Best Pr. Res Clin. Gastroenterol. 2018, 36–37, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Gallia, G.L.; Johnson, E.M.; Khalili, K. Puralpha: A multifunctional single-stranded DNA- and RNA-binding protein. Nucleic Acids Res. 2000, 28, 3197–3205. [Google Scholar] [CrossRef]
- Tretiakova, A.; Gallia, G.L.; Shcherbik, N.; Jameson, B.; Johnson, E.M.; Amini, S.; Khalili, K. Association of Puralpha with RNAs homologous to 7 SL determines its binding ability to the myelin basic protein promoter DNA sequence. J. Biol. Chem. 1998, 273, 22241–22247. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.W.; Pejovic, T.; Najfeld, V.; Ward, D.C.; Johnson, E.M. Localization of PURA, the gene encoding the sequence-specific single-stranded-DNA-binding protein Pur alpha, to chromosome band 5q31. Cytogenet Cell Genet 1995, 71, 64–67. [Google Scholar] [CrossRef]
- Johnson, E.M.; Daniel, D.C.; Gordon, J. The pur protein family: Genetic and structural features in development and disease. J. Cell Physiol. 2013, 228, 930–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergemann, A.D.; Johnson, E.M. The HeLa Pur factor binds single-stranded DNA at a specific element conserved in gene flanking regions and origins of DNA replication. Mol. Cell. Biol. 1992, 12, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbinian, N.; White, M.K.; Khalili, K. Regulation of the Pur-alpha promoter by E2F-1. J. Cell Biochem. 2006, 99, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Dong, C. Gluconeogenesis in cancer: Function and regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Grasmann, G.; Smolle, E.; Olschewski, H.; Leithner, K. Gluconeogenesis in cancer cells—Repurposing of a starvation-induced metabolic pathway? Biochim. Biophys. Acta Rev. Cancer 2019, 1872, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Messeguer, X.; Escudero, R.; Farre, D.; Nunez, O.; Martinez, J.; Alba, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef] [PubMed]
- Shelley, C.S.; Teodoridis, J.M.; Park, H.; Farokhzad, O.C.; Bottinger, E.P.; Arnaout, M.A. During differentiation of the monocytic cell line U937, Pur alpha mediates induction of the CD11c beta 2 integrin gene promoter. J. Immunol. 2002, 168, 3887–3893. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, N.; Bharti, A.; Shelley, C.S. hnRNP-K and Pur(alpha) act together to repress the transcriptional activity of the CD43 gene promoter. Blood 2002, 100, 3536–3544. [Google Scholar] [CrossRef]
- Shelley, C.S.; Da Silva, N.; Teodoridis, J.M. During U937 monocytic differentiation repression of the CD43 gene promoter is mediated by the single-stranded DNA binding protein Pur alpha. Br. J. Haematol. 2001, 115, 159–166. [Google Scholar] [CrossRef]
- Hariharan, S.; Kelm, R.J.; Strauch, A.R. The Purα/Purβ Single-Strand DNA-Binding Proteins Attenuate Smooth-Muscle Actin Gene Transactivation in Myofibroblasts. J. Cell. Physiol. 2014, 229, 1256–1271. [Google Scholar] [CrossRef]
- Mayorga, L.; Gamboni, B.; Mampel, A.; Roque, M. A frame-shift deletion in the PURA gene associates with a new clinical finding: Hypoglycorrhachia. Is GLUT1 a new PURA target? Mol. Genet Metab. 2018, 123, 331–336. [Google Scholar] [CrossRef]
- Liu, M.X.; Jin, L.; Sun, S.J.; Liu, P.; Feng, X.; Cheng, Z.L.; Liu, W.R.; Guan, K.L.; Shi, Y.H.; Yuan, H.X.; et al. Metabolic reprogramming by PCK1 promotes TCA cataplerosis, oxidative stress and apoptosis in liver cancer cells and suppresses hepatocellular carcinoma. Oncogene 2018, 37, 1637–1653. [Google Scholar] [CrossRef]
- Zhao, J.; Li, J.; Fan, T.W.M.; Hou, S.X. Glycolytic reprogramming through PCK2 regulates tumor initiation of prostate cancer cells. Oncotarget 2017, 8, 83602–83618. [Google Scholar] [CrossRef]
- Luo, S.; Li, Y.; Ma, R.; Liu, J.; Xu, P.; Zhang, H.; Tang, K.; Ma, J.; Liu, N.; Zhang, Y.; et al. Downregulation of PCK2 remodels tricarboxylic acid cycle in tumor-repopulating cells of melanoma. Oncogene 2017, 36, 3609–3617. [Google Scholar] [CrossRef] [PubMed]
- Leithner, K.; Hrzenjak, A.; Trotzmuller, M.; Moustafa, T.; Kofeler, H.C.; Wohlkoenig, C.; Stacher, E.; Lindenmann, J.; Harris, A.L.; Olschewski, A.; et al. PCK2 activation mediates an adaptive response to glucose depletion in lung cancer. Oncogene 2015, 34, 1044–1050. [Google Scholar] [CrossRef]
- Chun, S.Y.; Johnson, C.; Washburn, J.G.; Cruz-Correa, M.R.; Dang, D.T.; Dang, L.H. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Mol. Cancer 2010, 9, 293. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.; Yamasaki, T.; Shinohata, R.; Hata, M.; Nakajima, H.; Kono, N. Cloning and reporter analysis of human mitochondrial phosphoenolpyruvate carboxykinase gene promoter. Gene 2004, 338, 157–162. [Google Scholar] [CrossRef]
- Stark, R.; Kibbey, R.G. The mitochondrial isoform of phosphoenolpyruvate carboxykinase (PEPCK-M) and glucose homeostasis: Has it been overlooked? Biochim. Biophys. Acta 2014, 1840, 1313–1330. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Lucas, A.; Hyrossova, P.; Novellasdemunt, L.; Vinals, F.; Perales, J.C. Mitochondrial phosphoenolpyruvate carboxykinase (PEPCK-M) is a pro-survival, endoplasmic reticulum (ER) stress response gene involved in tumor cell adaptation to nutrient availability. J. Biol. Chem. 2014, 289, 22090–22102. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.; Yu, Y.; Zhu, L.; Xu, P.; Chen, J.; Ma, J.; Zhang, H.; Fang, H.; Sun, W.; Zhou, L.; et al. Hypoxia-reprogrammed tricarboxylic acid cycle promotes the growth of human breast tumorigenic cells. Oncogene 2019, 38, 6970–6984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciacovelli, M.; Frezza, C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J. 2017, 284, 3132–3144. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5’→3’) |
|---|---|
| Human PCK2-F | GGGCACTACCTGGAACACTG |
| Human PCK2-R | GCACTGTCCTCCCCCT-CTAA |
| Human PURA-F | TACGGCGTGTTTATGCGAGT |
| Human PURA-R | TTGCAGAAGGTGTGTCCGAA |
| Human β-actin-F | CATGTACGTTGCTATCCAGGC |
| Human β-actin-R | CTCCTTAATGTCAC-GCACGAT |
| Primer Name | Sequence (5’→3’) |
|---|---|
| PCK2-C1-F | GCTCACGCCTGTAAATTCCAG |
| PCK2-C1-R | TAGCTGGAATTACAGGCGC |
| PCK2-C2-F | TCCAGCTACTCTGCAGGTT |
| PCK2-C2-R | TCTCTCTACCCCTGGGTCT |
| PCK2-C3-F | CGTTCCTAGCTTGTTTGCCAC |
| PCK2-C3-R | CCAAACCGCCAAACCCATCT |
| CD11c-F | TCCATCTAAGCAAAGGGCATCA |
| CD11c-R | GCCAGGGGAAGGAAGAAGATT |
| Antibody | Source | Identifier | Dilution Multiple |
|---|---|---|---|
| PCK2-Rabbit antibody | Cell Signaling Technology | Cat# 6924S | 1:1000 |
| PURA-Rabbit antibody | Abcam | Cat# ab79936 | 1:1000 |
| β-actin-Mouse antibody | Boao rui jing | Cat# ab1015t | 1:5000 |
| Primer Name | Sequence (5’→3’) |
|---|---|
| PCK2-Luc-1500-227-F | CCGCTCGAGGTGTAGGCTAGAGCCTGAG |
| PCK2-Luc-1500-227-R | CCGCTCGAGGTGTAGGCTAGAGCCTGAG |
| PCK2-Luc-953-107-F | CCGCTCGAGCAGAAAAGGCGAGTGGATGC |
| PCK2-Luc-953-107-R | CCCAAGCTTTGGCACTAGCTTTCCTCCG |
| PCK2-Luc-221-107-F | CCGCTCGAGATGCCCAACCCCCTAACTTC |
| PCK2-Luc-221-107-R | CCCAAGCTTTGGCACTAGCTTTCCTCCG |
| PCK2-Luc-S1-MU-F | CCCTTTATTCTTAGTGTGCAGTGAGCCGAGATCG |
| PCK2-Luc-S1-MU-R | GCACACTAAGAATAAAGGGTTCAAGCAATTCTCCTGTCTCA |
| pCMV6-ATF4-F | CCCAAGCTTATGACCGAAATGAGCTTCCT |
| pCMV6-ATF4-R | CCGCTCGAGGGGGACCCTTTTCTTCCC |
| siRNA | Sequence (5’→3’) |
|---|---|
| siCtrl | UUCUCCGAACGUGUCACGU |
| siPURα-1 | CCACCUAUCGCAACUCCAUTT |
| siPURα-2 | CCAAGUUCGGACACACCUUTT |
| shNC | ACAGAAGCGATTGTTGATC |
| shPURα32 | CCACCAACTGACAGTTTCTCT |
| shPURα34 | AGCCGCCTTACTCTCTCCATG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Y.; Gao, J.; Jing, Z.; Zhao, Y.; Sun, Y.; Zhao, X. PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells. Genes 2020, 11, 1301. https://doi.org/10.3390/genes11111301
Sun Y, Gao J, Jing Z, Zhao Y, Sun Y, Zhao X. PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells. Genes. 2020; 11(11):1301. https://doi.org/10.3390/genes11111301
Chicago/Turabian StyleSun, Yan, Jiajia Gao, Zongpan Jing, Yan Zhao, Yulin Sun, and Xiaohang Zhao. 2020. "PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells" Genes 11, no. 11: 1301. https://doi.org/10.3390/genes11111301
APA StyleSun, Y., Gao, J., Jing, Z., Zhao, Y., Sun, Y., & Zhao, X. (2020). PURα Promotes the Transcriptional Activation of PCK2 in Oesophageal Squamous Cell Carcinoma Cells. Genes, 11(11), 1301. https://doi.org/10.3390/genes11111301