Expression Quantitative Trait Locus Mapping in Pulmonary Arterial Hypertension

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants and Sample Processing
2.2. Gene Expression Data
2.3. Genetic Data
2.4. eQTL Effect Estimation
2.5. Confirmation Rate
2.6. Overlap with Variants from the GWAS Catalog
2.7. Functional Enrichment Analysis of Genes with Novel eQTL
3. Results
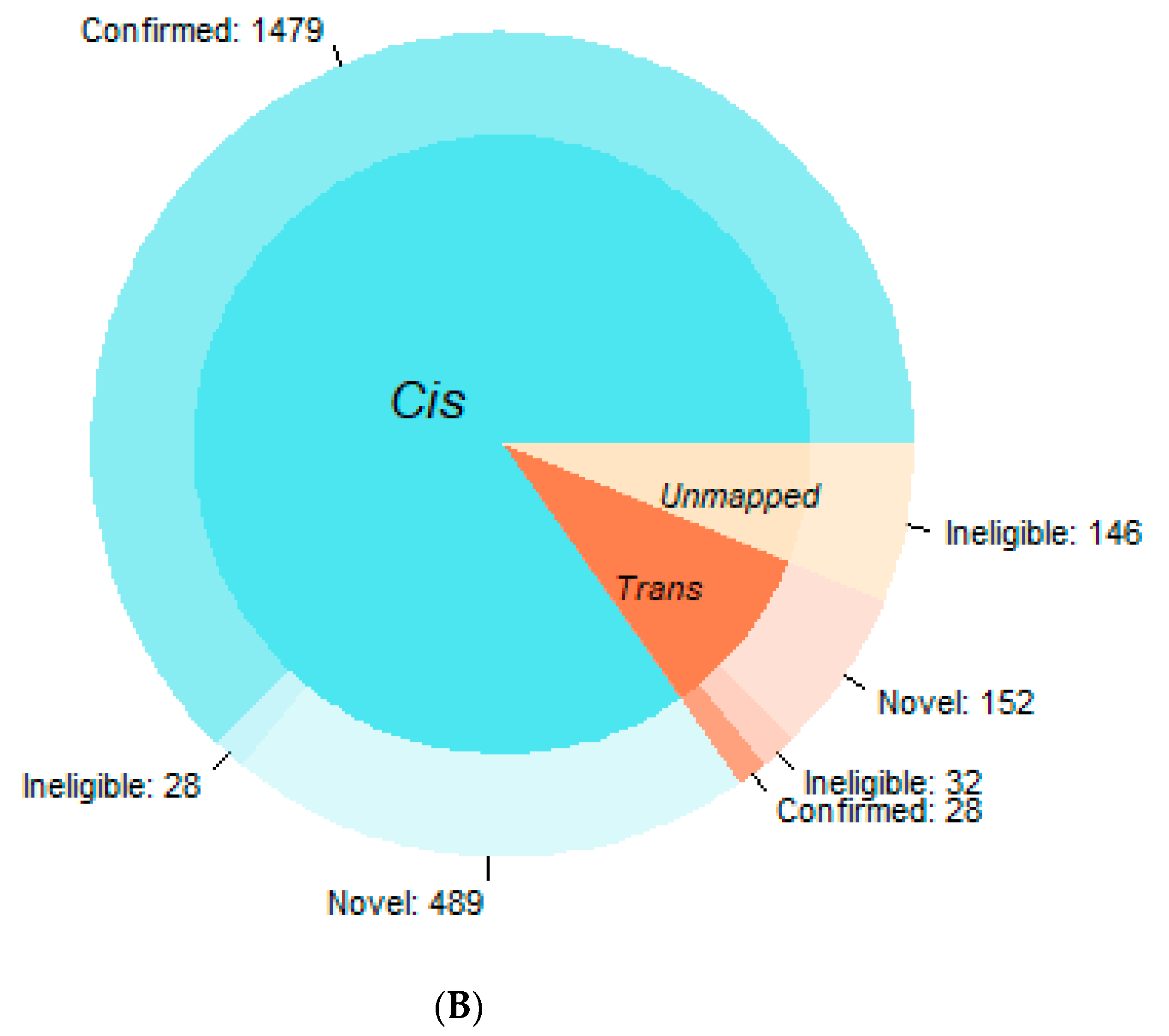
3.1. Confirmation Rate
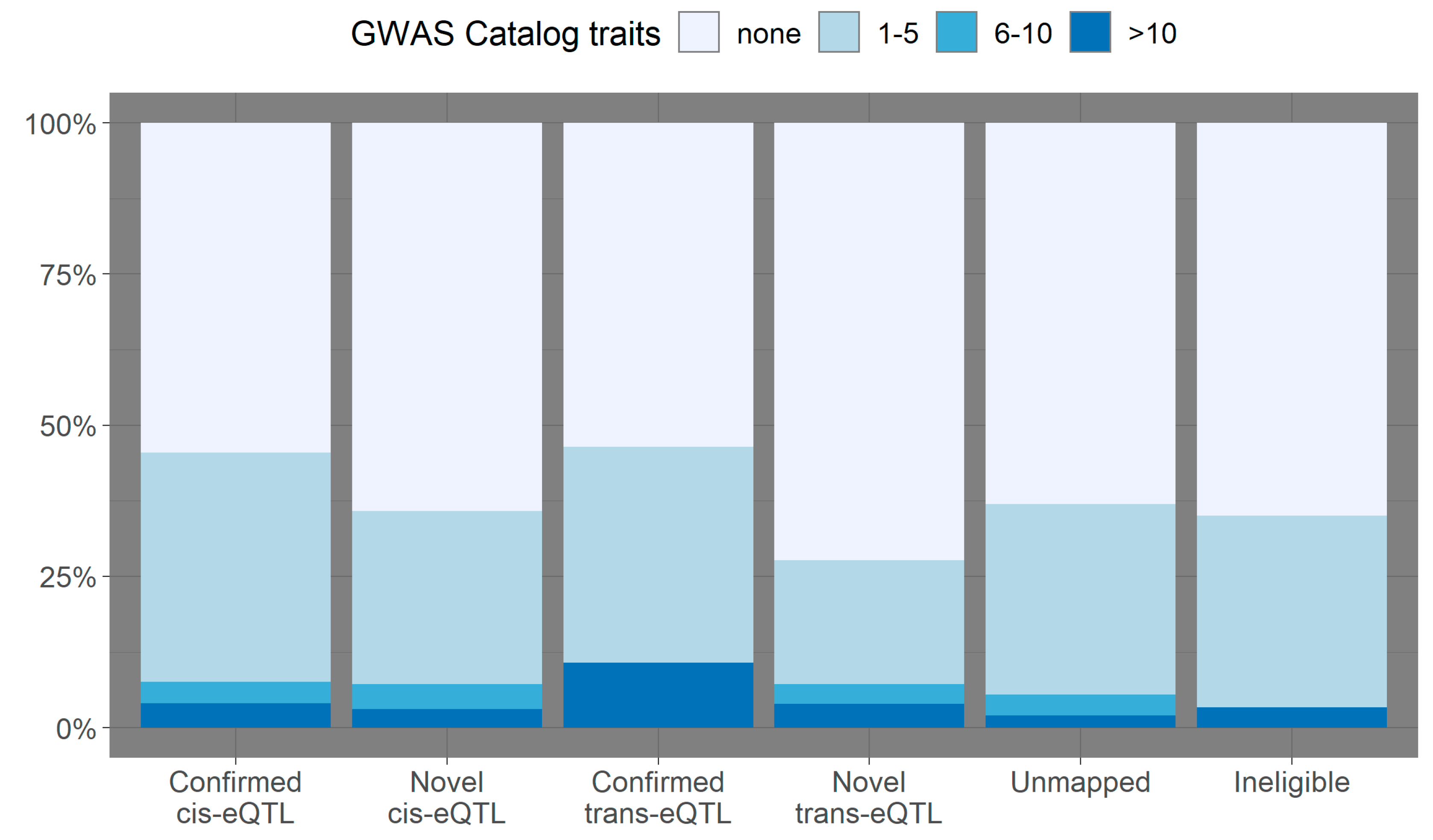
3.2. Overlap with Variants from the GWAS Catalog
3.3. Functional Enrichment Analysis of Genes with Novel eQTL
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Gamazon, E.R.; Segrè, A.V.; van de Bunt, M.; Wen, X.; Xi, H.S.; Hormozdiari, F.; Ongen, H.; Konkashbaev, A.; Derks, E.M.; Aguet, F.; et al. Using an atlas of gene regulation across 44 human tissues to inform complex disease- and trait-associated variation. Nat. Genet. 2018, 50, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Westra, H.J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Joehanes, R.; Zhang, X.; Huan, T.; Yao, C.; Ying, S.X.; Nguyen, Q.T.; Demirkale, C.Y.; Feolo, M.L.; Sharopova, N.R.; Sturcke, A.; et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, D.V.; Deelen, P.; Vermaat, M.; van Iterson, M.; van Galen, M.; Arindrarto, W.; van ‘t Hof, P.; Mei, H.; van Dijk, F.; Westra, H.J.; et al. Identification of context-dependent expression quantitative trait loci in whole blood. Nat. Genet. 2017, 49, 139–145. [Google Scholar] [CrossRef]
- Gonorazky, H.D.; Naumenko, S.; Ramani, A.K.; Nelakuditi, V.; Mashouri, P.; Wang, P.; Kao, D.; Ohri, K.; Viththiyapaskaran, S.; Tarnopolsky, M.A.; et al. Expanding the Boundaries of RNA Sequencing as a Diagnostic Tool for Rare Mendelian Disease. Am. J. Hum. Genet. 2019, 104, 466–483. [Google Scholar] [CrossRef]
- Dancik, G.M. An online tool for evaluating diagnostic and prognostic gene expression biomarkers in bladder cancer. BMC Urol. 2015, 15, 59. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.U.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- VanderWeele, T.J.; Tchetgen Tchetgen, E.J.; Cornelis, M.; Kraft, P. Methodological challenges in mendelian randomization. Epidemiology 2014, 25, 427–435. [Google Scholar] [CrossRef]
- Singh, A.J.; Ramsey, S.A.; Filtz, T.M.; Kioussi, C. Differential gene regulatory networks in development and disease. Cell. Mol. Life Sci. 2018, 75, 1013–1025. [Google Scholar] [CrossRef]
- Gov, E.; Arga, K.Y. Differential co-expression analysis reveals a novel prognostic gene module in ovarian cancer. Sci. Rep. 2017, 7, 4996. [Google Scholar] [CrossRef]
- Zhu, L.; Ding, Y.; Chen, C.-Y.; Wang, L.; Huo, Z.; Kim, S.; Sotiriou, C.; Oesterreich, S.; Tseng, G.C. MetaDCN: Meta-analysis framework for differential co-expression network detection with an application in breast cancer. Bioinformatics 2016, 33, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Chen, M.; Zhang, C.K.; Cho, J.; Zhao, H. Guilt by rewiring: Gene prioritization through network rewiring in genome wide association studies. Hum. Mol. Genet. 2014, 23, 2780–2790. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Groves, B.; Badesch, D.B.; Voelkel, N.F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 1994, 144, 275–285. [Google Scholar] [PubMed]
- Galie, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar] [PubMed]
- Rhodes, C.J.; Batai, K.; Bleda, M.; Haimel, M.; Southgate, L.; Germain, M.; Pauciulo, M.W.; Hadinnapola, C.; Aman, J.; Girerd, B.; et al. Genetic determinants of risk in pulmonary arterial hypertension: International genome-wide association studies and meta-analysis. Lancet Respir. Med. 2019, 7, 227–238. [Google Scholar] [CrossRef]
- Graf, S.; Haimel, M.; Bleda, M.; Hadinnapola, C.; Southgate, L.; Li, W.; Hodgson, J.; Liu, B.; Salmon, R.M.; Southwood, M.; et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 2018, 9, 1416. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Otero-Nunez, P.; Wharton, J.; Swietlik, E.M.; Kariotis, S.; Harbaum, L.; Dunning, M.J.; Elinoff, J.M.; Errington, N.; Thompson, A.A.R.; et al. Whole-Blood RNA Profiles Associated with Pulmonary Arterial Hypertension and Clinical Outcome. Am. J. Respir. Crit. Care Med. 2020, 202, 586–594. [Google Scholar] [CrossRef]
- Finotello, F.; Mayer, C.; Plattner, C.; Laschober, G.; Rieder, D.; Hackl, H.; Krogsdam, A.; Loncova, Z.; Posch, W.; Wilflingseder, D.; et al. Molecular and pharmacological modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med. 2019, 11, 34. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 25. [Google Scholar] [CrossRef]
- Band, G.; Marchini, J. Qctool. Available online: https://www.well.ox.ac.uk/~gav/qctool/index.html (accessed on 10 September 2020).
- QUICKTEST. Available online: https://wp.unil.ch/sgg/program/quicktest/ (accessed on 10 September 2020).
- Aguet, F.; Barbeira, A.N.; Bonazzola, R.; Brown, A.; Castel, S.E.; Jo, B.; Kasela, S.; Kim-Hellmuth, S.; Liang, Y.; Oliva, M.; et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. bioRxiv 2019, 787903v1. [Google Scholar] [CrossRef]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 8, D1005–D1012. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [PubMed]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten Years of Pathway Analysis: Current Approaches and Outstanding Challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36, D480–D484. [Google Scholar] [CrossRef]
- Powell, J.E.; Henders, A.K.; McRae, A.F.; Wright, M.J.; Martin, N.G.; Dermitzakis, E.T.; Montgomery, G.W.; Visscher, P.M. Genetic control of gene expression in whole blood and lymphoblastoid cell lines is largely independent. Genome Res. 2012, 22, 456–466. [Google Scholar] [CrossRef]
- Barreiro, L.B.; Tailleux, L.; Pai, A.A.; Gicquel, B.; Marioni, J.C.; Gilad, Y. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 2012, 109, 1204. [Google Scholar] [CrossRef]
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; .McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef]
- Çalışkan, M.; Baker, S.W.; Gilad, Y.; Ober, C. Host Genetic Variation Influences Gene Expression Response to Rhinovirus Infection. PLoS Genet. 2015, 11, e1005111. [Google Scholar] [CrossRef]
- Zhu, Z.; Wang, X.; Li, X.; Lin, Y.; Shen, S.; Liu, C.L.; Hobbs, B.D.; Hasegawa, K.; Liang, L.; Boezen, H.M.; et al. Genetic overlap of chronic obstructive pulmonary disease and cardiovascular disease-related traits: A large-scale genome-wide cross-trait analysis. Respir. Res. 2019, 20, 64. [Google Scholar] [CrossRef]
- Kichaev, G.; Bhatia, G.; Loh, P.-R.; Gazal, S.; Burch, K.; Freund, M.K.; Schoech, A.; Pasaniuc, B.; Price, A.L. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am. J. Hum. Genet. 2019, 104, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Evangelou, E.; Warren, H.R.; Mosen-Ansorena, D.; Mifsud, B.; Pazoki, R.; Gao, H.; Ntritsos, G.; Dimou, N.; Cabrera, C.P.; Karaman, I.; et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018, 50, 1412–1425. [Google Scholar] [CrossRef] [PubMed]
- Merla, G.; Brunetti-Pierri, N.; Piccolo, P.; Micale, L.; Loviglio Maria, N. Supravalvular Aortic Stenosis. Circul. Cardiovasc. Genet. 2012, 5, 692–696. [Google Scholar] [CrossRef]
- Rabinovitch, M.; Guignabert, C.; Humbert, M.; Nicolls, M.R. Inflammation and Immunity in the Pathogenesis of Pulmonary Arterial Hypertension. Circul. Res. 2014, 115, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Graham, B. How does inflammation contribute to pulmonary hypertension? Eur. Respir. J. 2018, 51, 1702403. [Google Scholar] [CrossRef]
- Andiappan, A.K.; Melchiotti, R.; Poh, T.Y.; Nah, M.; Puan, K.J.; Vigano, E.; Haase, D.; Yusof, N.; San Luis, B.; Lum, J.; et al. Genome-wide analysis of the genetic regulation of gene expression in human neutrophils. Nat. Commun. 2015, 6, 7971. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Westra et al. | Joehanes et al. | GTEx | This Study | |
|---|---|---|---|---|
| Gene expression array | Illumina HumanHT-12 v4.0 | Affymetrix HuEx 1.0 ST | RNAseq | RNAseq |
| Genotyping panel | HapMap2 | 1000-Genomes | WGS | WGS |
| MAF threshold | ≥5% | ≥1% | ≥1% | ≥5% |
| n variants in analysis | not reported | 8,510,936 | 10,008,325 | 7,362,566 |
| cis-eQTL | ≤250 kb from PMP | ≤1 Mb from the TSS | ≤1 Mb from the TSS | ≤1 Mb from the TSS |
| trans-eQTL | ≥5 Mb from PMP | >1 Mb from the TSS | >1 Mb from the TSS | >1 Mb from the TSS |
| Gene Set | Description | Size | Overlap | Expectation | Enrichment Ratio | p-Value | FDR |
|---|---|---|---|---|---|---|---|
| hsa04742 | Taste transduction | 48 | 8 | 0.87 | 9.24 | 1.78 × 10−6 | 5.72 × 10−4 |
| hsa05332 | Graft-versus-host disease | 32 | 6 | 0.58 | 10.40 | 1.85 × 10−5 | 2.86 × 10−3 |
| hsa05320 | Autoimmune thyroid disease | 34 | 6 | 0.61 | 9.79 | 2.66 × 10−5 | 2.86 × 10−3 |
| hsa05330 | Allograft rejection | 33 | 5 | 0.59 | 8.40 | 2.75 × 10−4 | 0.02 |
| hsa04940 | Type I diabetes mellitus | 36 | 5 | 0.65 | 7.70 | 4.19 × 10−4 | 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulrich, A.; Otero-Núñez, P.; Wharton, J.; Swietlik, E.M.; Gräf, S.; Morrell, N.W.; Wang, D.; Lawrie, A.; Wilkins, M.R.; Prokopenko, I.; et al. Expression Quantitative Trait Locus Mapping in Pulmonary Arterial Hypertension. Genes 2020, 11, 1247. https://doi.org/10.3390/genes11111247
Ulrich A, Otero-Núñez P, Wharton J, Swietlik EM, Gräf S, Morrell NW, Wang D, Lawrie A, Wilkins MR, Prokopenko I, et al. Expression Quantitative Trait Locus Mapping in Pulmonary Arterial Hypertension. Genes. 2020; 11(11):1247. https://doi.org/10.3390/genes11111247
Chicago/Turabian StyleUlrich, Anna, Pablo Otero-Núñez, John Wharton, Emilia M. Swietlik, Stefan Gräf, Nicholas W. Morrell, Dennis Wang, Allan Lawrie, Martin R. Wilkins, Inga Prokopenko, and et al. 2020. "Expression Quantitative Trait Locus Mapping in Pulmonary Arterial Hypertension" Genes 11, no. 11: 1247. https://doi.org/10.3390/genes11111247
APA StyleUlrich, A., Otero-Núñez, P., Wharton, J., Swietlik, E. M., Gräf, S., Morrell, N. W., Wang, D., Lawrie, A., Wilkins, M. R., Prokopenko, I., Rhodes, C. J., on behalf of The NIHR BioResource—Rare Diseases Consortium, & UK PAH Cohort Study Consortium. (2020). Expression Quantitative Trait Locus Mapping in Pulmonary Arterial Hypertension. Genes, 11(11), 1247. https://doi.org/10.3390/genes11111247