Clinical Presentation of the c.3844T>C (p.Trp1282Arg, W1282R) Variant in Russian Cystic Fibrosis Patients

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
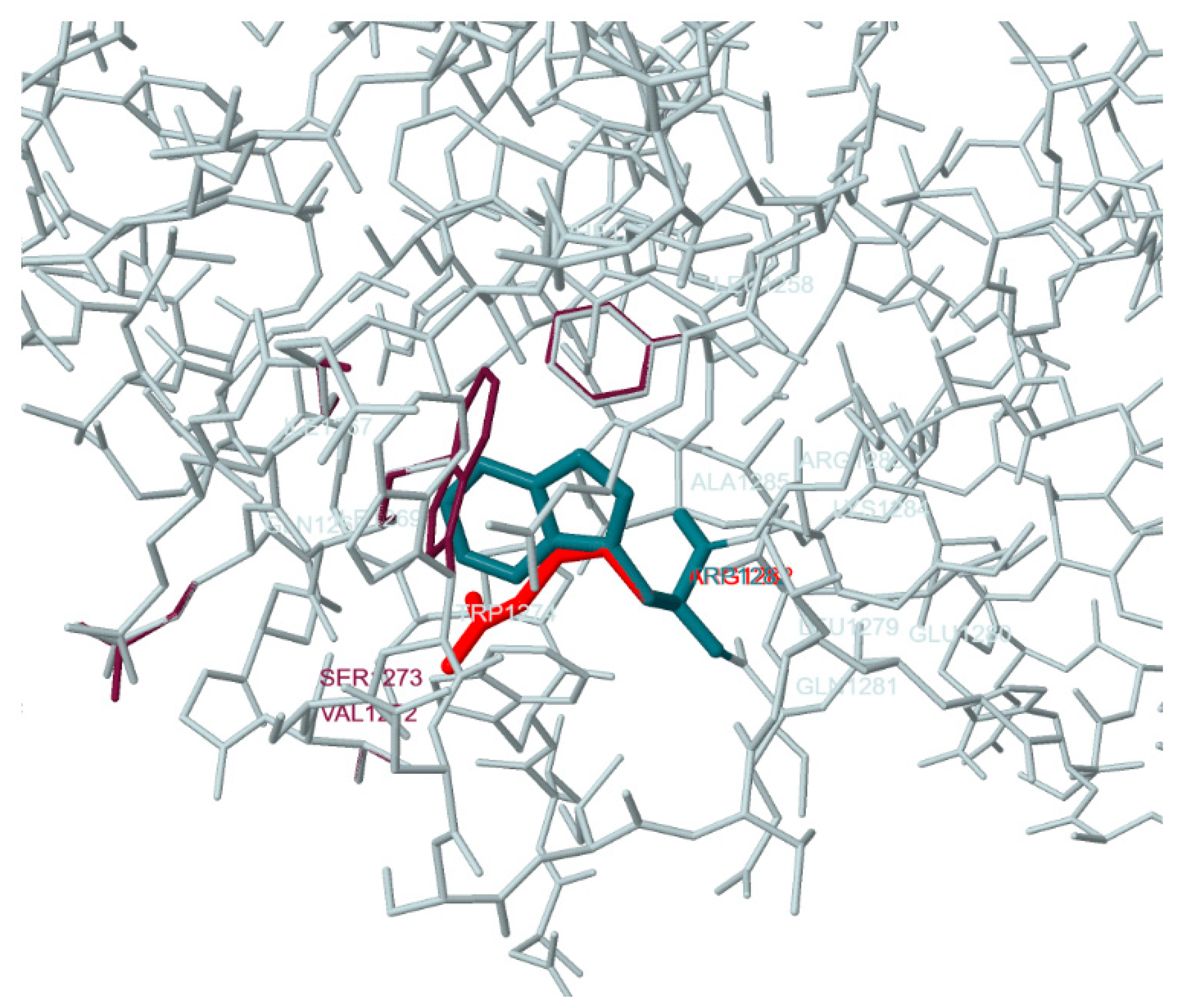
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sosnay, P.R.; Raraigh, K.S.; Gibson, R.L. Molecular Genetics of Cystic Fibrosis Transmembrane Conductance Regulator: Genotype and Phenotype. Pediatr. Clin. N. Am. 2016, 63, 585–598. [Google Scholar] [CrossRef] [PubMed]
- Farrell, P.M.; White, T.B.; Howenstine, M.S.; Munck, A.; Parad, R.B.; Rosenfeld, M.; Sommerburg, O.; Accurso, F.J.; Davies, J.C.; Rock, M.J.; et al. Diagnosis of Cystic Fibrosis in Screened Populations. J. Pediatr. 2017, 181S, S33–S44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Cystic Fibrosis Mutation Database. Available online: http://www.genet.sickkids.on.ca (accessed on 9 April 2020).
- CFTR2. Clinical and Functional Translation of CFTR. Available online: https://www.cftr2.org/ (accessed on 9 April 2020).
- Krasovskiy, S.A.; Kashirskaya, N.Y.; Chernyak, A.V.; Amelina, E.L.; Petrova, N.V.; Polyakov, A.V.; Kondratyeva, E.I.; Voronkova, A.Y.; Usacheva, M.V.; Adyan, T.A.; et al. Genetic characteristic of CF patients in Russian Federation according to National Registre (2014). Pulmonologiya 2016, 26, 133–151. (In Russian) [Google Scholar] [CrossRef] [Green Version]
- Register of Patients with Cystic Fibrosis in the Russian Federation 2018; Amelina, E.A.; Kashirskaya, N.Y.; Kondratieva, E.I.; Krasovsky, S.A.; Starinova, M.A.; Voronkova, A.Y.-M. (Eds.) Izdatelskiy Dom “MEDPRAKTIKA M”: Moscow, Russia, 2020; 68p, (In Russian). ISBN 978-5-98803-432-2. [Google Scholar]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilieva, T.A.; Timkovskaya, E.E.; Voronkova, A.Y.; Shabalova, L.A.; Kondrateva, E.I.; Sherman, V.D.; Kapranov, N.I.; Zinchenko, R.A.; et al. High proportion of W1282X mutation in CF patients from Karachai-Cherkessia. J. Cyst. Fibros. 2016, 15, e28–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, N.V.; Kashirskaya, N.Y.; Saydaeva, D.K.; Polyakov, A.V.; Adyan, T.A.; Simonova, O.I.; Gorinova, Y.V.; Kondratyeva, E.I.; Sherman, V.D.; Novoselova, O.G.; et al. Spectrum of CFTR mutations in Chechen cystic fibrosis patients: High frequency of c.1545_1546delTA (p.Tyr515X; 1677delTA) and c.274G>A (p.Glu92Lys, E92K) mutations in North Caucasus. BMC Med. Genet. 2019, 20, 44. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.A.; Polyakov, A.V.; Abrukova, A.V.; Savaskina, E.N. Mutation p.E92K is the primary cause of cystic fibrosis in Chuvashes. Russ. J. Genet. 2012, 48, 731–737. [Google Scholar] [CrossRef]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Kondratyeva, E.I.; Zhekaite, E.K.; Voronkova, A.Y.; Sherman, V.A.; Galkina, G.E.K.; Kutsev, S.I.; Marakhonov, A.V.; et al. Analysis of CFTR mutation spectrum in ethnic Russian Cystic Fibrosis patients. Genes 2020, 11, 554. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, J.L.; Macek, M., Jr.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- WHO Human Genetics Programme. The Molecular Genetic Epidemiology of Cystic Fibrosis; Report of a Joint Meeting of WHO/ECFTN/ICF(M)/ECFS/World Health Organization (WHO); WHO: Genoa, Italy, 2002; Available online: https://www.cfww.org/docs/who/2002/who_hgn_cf_wg_04.02.pdf (accessed on 8 February 2020).
- Petrova, N.V.; Marakhonov, A.V.; Vasilyeva, T.A.; Kashirskaya, N.Y.; Ginter, E.K.; Kutsev, S.I.; Zinchenko, R.A. Comprehensive genotyping reveals novel CFTR variants in cystic fibrosis patients from the Russian Federation. Clin. Genet. 2018, 95, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Voronkova, A.Y.; Kondratyeva, E.I.; Sherman, V.D.; Novoselova, O.G.; Krasovsky, S.А.; Chernyak, А.V.; Amelina, E.L.; et al. Phenotypic features in Russian CF patients with L138ins (p.Leu138dup) mutation. Pediatriya 2017, 96, 64–72. (In Russian) [Google Scholar] [CrossRef]
- Krasovsky, S.A.; Petrova, N.V.; Stepanova, A.A.; Usacheva, M.V.; Samoilenko, V.A.; Amelina, E.L.; Nikonova, V.S. Clinical course of cystic fibrosis on adult patients carrying “mild” mutations. Pulmonologiya 2012, 6, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell. 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechecchi, M.C.; Tamanini, A.; Cabrini, G. Molecular basis of cystic fibrosis: From bench to bedside. Ann. Transl. Med. 2018, 6, 334. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Kondratyeva, E.I.; Krasovsky, S.A.; Polyakov, A.V.; Ivachshenko, T.E.; Pavlov, A.E.; Zinchenko, R.A.; Ginter, E.K.; Kutsev, S.I.; Odinokova, O.N.; et al. National Consensus Project «Cystic fibrosis: Definition, diagnostic criteria, treatment» Section «Genetics of Cystic Fibrosis. Molecular genetic diagnosis of cystic fibrosis». Meditsinskaya Genetica 2016, 11, 29–45. (In Russian) [Google Scholar]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human UNIT 7.20 Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Ittisoponpisan, S.; Islam, S.A.; Khanna, T.; Alhuzimi, E.; David, A.; Sternberg, M.J.E. Can predicted protein 3D structures provide reliable insights into whether missense variants are disease associated? J. Mol. Biol. 2019, 431, 2197–2212. [Google Scholar] [CrossRef] [PubMed]
- Ivaschenko, T.E.; Baranov, V.S.; Dean, M. Two new mutations detected by single-strand conformation polymorphism analysis in cystic fibrosis from Russia. Hum. Genet. 1993, 91, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Krasovsky, S.A.; Amelina, E.L.; Usacheva, M.V.; Stepanova, A.A.; Polyakov, A.V.; Ferec, C.; Audrezete, M.P. Phenotypic features of W1282 mutation of cystic fibrosis regulator protein. In Proceedings of the XXIII National Congress on Respiratory Diseases; Designpress: Moscow, Russia, 2013; 528p, Available online: https://spulmo.ru/download/Tezisi_2013(23).pdf (accessed on 9 April 2020). (In Russian)
- National Center for Biotechnology Information (NCBI-ClinVar). Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 9 April 2020).
- ExAC Browser (Beta). Exome Aggregation Consortium. Available online: http://exac.broadinstitute.org/ (accessed on 9 April 2020).
- Castellani, C.; Cuppens, H.; Macek, M., Jr.; Cassiman, J.J.; Kerem, E.; Durie, P.; Tullis, E.; Assael, B.M.; Bombieri, C.; Brown, A.; et al. Consensus on the use and interpretation of cystic fibrosis mutation analysis in clinical practice. J. Cyst. Fibros. 2008, 7, 179–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Functional Class | Legacy Name | cDNA Name | Protein Name | No. of Patients |
|---|---|---|---|---|
| II | F508del | c.1521_1523delCTT | p.Phe508del | 19 |
| I | CFTRdele2,3 | c.54-5940_273+10250del21kb | p.Ser18Arg*fsX16 | 4 |
| V | 3849+10kbC-T | c.3718-2477C>T | p.spl | |
| I | 580-1G>T | c.712-1G->T | p.spl | 2 |
| IV | R334W | c.1000C>T | p.Arg334Trp | 2 |
| I | W1282X | c.3846G>A | p.Trp1282X | 2 |
| II-III | E92K | c.274G>A | p.Glu92Lys | 2 |
| I | 2184insA | c.2052_2053insA | p.Gln685ThrfsX4 | 1 |
| I | 3821delT | c.3691delT | p.Ser1231ProfsX4 | 1 |
| n.d. 1 | 4382delA | c.4251delA | p.Glu1418ArgfsX14 | 1 |
| I | 1898+1G>A | c.1766+1G>A | p.spl | 1 |
| I | 541delC | c.409delC | p.Leu137SerfsX16 | 1 |
| III | D1152H | c.3454G>C | p.Asp1152His | 1 |
| n.d. 1 | L138ins | c.413_415dupTAC | p.Leu138dup | 1 |
| II | R1066C | c.3196C>T | p.Arg1066Cys | 1 |
| III | L1335P | c.4004T>C | p.Leu1335Pro | 1 |
| Mean Age (Years) | Groups (Genotypes) | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| at last visit to clinic | 13.48 ± 1.67 (0.73–30.71); 31 | 16.18 ± 1.23 (0.60–32.60); 50 | 12.94 ± 0.90 (0.53–31.07); 100 | 23.44 ± 6.67 (0.74–60.06); 9 |
| p = 0.224 | p = 0.314 | p = 0.23 | ||
| at diagnosis | 2.74 ± 0.88 (0.075–23.00); 31 | 9.01 ± 1.06 (0.10–27.78); 50 | 2.24 ± 0.36 (0.00–15.35); 99 | 13.07 ± 5.09 (0.12–39.60); 9 |
| p = 0.000019 | p = 0.437 | p = 0.052 | ||
| at diagnosis before neonatal screening | 3.98 ± 1.46 (0.19–23.00); 17 | 11.40 ± 1.18 (0.63–27.78); 37 | 3.65 ± 0.57 (0.00–15.35); 55 | 19.50 ± 6.11 (4.49–39.60); 6 |
| p = 0.00033 | p = 0.31 | p = 0.0033 | ||
| at diagnosis by neonatal screening | 0.27 ± 0.13 (0.075–1.51); 11 | 2.38 ± 0.70 (0.10–7.52); 12 | 0.46 ± 0.13 (0.008–3.88); 44 | 0.20 ± 0.05 (0.11–0.29); 3 |
| p = 0.015 | p = 0.37 | p = 0.35 | ||
| with chronic Ps. aeruginosa | 17.76 ± 2.38 (1.10–30.71); 15 | 19.55 ± 1.43 (2.88–32.60); 26 | 16.14 ± 1.39 (1.38–30.97); 38 | 32.16 ± 8.10 (13.0–60.06); 5 |
| p = 0.64 | p = 0.47 | p = 0.088 | ||
| without chronic Ps. aeruginosa | 9.47 ± 1.92 (0.73–23.00); 16 | 11.82 ± 1.72 (0.60–29.06); 23 | 10.78 ± 1.12 (0.53–31.07); 61 | 12.54 ± 9.29 (0.74–40.24); 4 |
| p = 0.12 | p = 0.70 | p = 0.71 | ||
| respiratory function (from sample) | 17.45 ± 1.64 (6.19–29.78); 18 | 18.31 ± 1.41 (6.73–32.60); 24 | 16.85 ± 0.88 (5.12–30.97); 54 | 29.59 ± 6.91 (6.14–60.06); 7 |
| p = 0.56 | p = 0.86 | p = 0.13 | ||
| respiratory function (total from RCFPR) | 17.45 ± 1.64 (6.19–29.78); 18 | 25.22 ± 1.61 (6.73–49.89); 42 | 14.39 ± 0.34 (5.03–42.35); 430 | |
| p = 0.0067 | p = 0.051 | |||
| Mean Values | Groups (Genotypes) | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| Sweat chlorides (mmol/L) | 106.82 ± 4.38 (62.0–160.0); 29 | 80.84 ± 3.22 (52.0–117.0); 40 | 105.02 ± 2.12 (50.0–157.0); 91 | 104.00 ± 20.46 (52.0–160.0); 5 |
| p = 0.000031 | p = 0.78 | p = 1.00 | ||
| BMI (all) (kg/m2) | 16.41 ± 0.53 (12.24–26.20); 30 | 16.79 ± 0.33 (13.00–22.00); 50 | 16.61 ± 0.25 (11.80–24.80); 96 | 17.60 ± 1.36 (13.15–23.88); 8 |
| p = 0.39 | p = 0.53 | p = 0.56 | ||
| BMI (children) (kg/m2) | 15.42 ± 0.52 (12.24–20.20); 20 | 16.32 ± 0.43 (13.00–22.00); 25 | 15.98 ± 0.25 (11.80–21.60); 73 | 15.61 ± 0.26 (15.31–16.44); 3 |
| p = 0.14 | p = 0.29 | p = 0.53 | ||
| BMI (adults) (kg/m2) | 18.37 ± 0.97 (15.80–26.20); 10 | 17.26 ± 0.49 (13.30–21.70); 25 | 18.57 ± 0.52 (14.90–24.80); 19 | 18.74 ± 2.06 (13.15–23.88); 5 |
| p = 0.43 | p = 0.31 | p = 0.90 | ||
| FEV1 (% predicted) | 64.54 ± 7.01 (16.76–102.50); 18 | 54.35 ± 4.32 (21.60–86.30); 24 | 79.64 ± 3.72 (27.20–142.48); 54 | 62.13 ± 13.41 (17.60–105.30); 7 |
| p = 0.35 | p = 0.095 | p = 0.71 | ||
| FEV1 (% predicted) (total from RCFPR) | 64.54 ± 7.01 (16.76–102.50); 18 | 54.05 ± 3.29 (18.30–87.90); 42 | 76.93 ± 1.15 (15.00–142.48); 430 | |
| p = 0.23 | p = 0.064 | |||
| FVC (% predicted) | 76.79 ± 4.66 (40.38–117.00); 18 | 69.27 ± 4.96 (22.70–105.80); 24 | 89.69 ± 3.14 (43.30–148.35); 53 | 69.01 ± 10.47 (26.10–102.20); 7 |
| p = 0.32 | p = 0.026 | p = 0.69 | ||
| FVC (% predicted) (total from RCFPR) | 64.54 ± 7.01 (16.76–102.50); 18 | 70.38 ± 3.71 (21.80–106.00); 42 | 86.20 ± 1.02 (24.70–176.20); 427 | |
| p = 0.21 | p = 0.047 | |||
| Clinical Features | Groups | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||||||
| n/N | % | n/N | % | n/N | % | n/N | % | ||
| normal RF (FVC&FEV1 > 80% pr.) | 4/18 | 22.2 | 2/24 | 8.3 | 29/54 | 53.7 | 3/7 | 42.8 | |
| p = 0.37 | p = 0.0286 | p = 0.355 | |||||||
| First or second-degree decrease of RF (FVC&FEV1 40–79.9% pr.) | 9/18 | 50.0 | 16/24 | 66.7 | 21/54 | 38.9 | 2/7 | 28.6 | |
| p = 0.34 | p = 0.42 | p = 0.231 | |||||||
| Third-degree decrease of RF (FVC&FEV1 < 40% pr.) | 5/18 | 27.8 | 6/24 | 25.0 | 4/54 | 7.4 | 2/7 | 28.6 | |
| p = 1.00 | p = 0.0378 | p = 1.00 | |||||||
| Fecal elastase 1 | >200 µg/g | 2 | 14.3 | 16 | 88.9 | 4 | 8.9 | 3 | 60.0 |
| <200 µg/g | 12 | 85.7 | 2 | 11.1 | 41 | 91.1 | 2 | 40.0 | |
| p = 0.0001 | p = 0.62 | p = 0.084 | |||||||
| Microflora in anamnesis | S. aureus | 16/31 | 51.6 | 20/49 | 40.8 | 53/100 | 53.0 | 2/9 | 22.2 |
| p = 0.36 | p = 1.00 | p = 0.149 | |||||||
| Non-tuberculous Mycobacteria (NTM) | 0/22 | 0.0 | 3/35 | 8.6 | 1/74 | 1.4 | 0/7 | 0.0 | |
| p = 0.28 | p = 1.00 | ||||||||
| Achromobacter | 0/30 | 0.0 | 2/48 | 4.2 | 8/96 | 8.3 | 1/9 | 11.1 | |
| p = 0.52 | p = 0.19 | p = 0.23 | |||||||
| St. maltophilia | 1/31 | 3.2 | 1/49 | 2.0 | 3/99 | 3.0 | 0/9 | 0.0 | |
| p = 1.00 | p = 1.00 | p = 1.00 | |||||||
| B. cepacia | 5/31 | 16.1 | 3/49 | 6.1 | 7/100 | 7.0 | 1/9 | 11.1 | |
| p = 0.25 | p = 0.14 | p = 1.00 | |||||||
| Gram-negative flora | 4/30 | 13.3 | 3/50 | 6.0 | 10/100 | 10.0 | 1/9 | 11.1 | |
| p = 0.42 | p = 0.73 | p = 1.00 | |||||||
| p. aeruginosa | chronic | 15/31 | 48.4 | 26/49 | 53.1 | 38/99 | 38.4 | 5/9 | 55.6 |
| p = 0.82 | p = 0.40 | p = 1.00 | |||||||
| chronic: children | 7/20 | 35.0 | 8/25 | 32.0 | 23/70 | 31.5 | 1/4 | 25.0 | |
| p = 1.00 | p = 1.00 | p = 1.00 | |||||||
| chronic: adults | 8/11 | 72.7 | 18/24 | 75.0 | 15/26 | 57.7 | 4/5 | 75.0 | |
| p = 1.00 | p = 0.476 | p = 1.00 | |||||||
| Allergic bronchopulmonary aspergillosis (ABPA) | 1/31 | 3.2 | 1/50 | 2.0 | 0/100 | 0.0 | 1/9 | 11.1 | |
| p = 1.00 | p = 0.236 | p = 0.403 | |||||||
| Nasal polyposis | 5/25 | 20.0 | 7/40 | 17.5 | 24/89 | 26.9 | 3/7 | 42.9 | |
| p = 1.00 | p = 0.61 | p = 0.326 | |||||||
| Meconium ileus | 3/31 | 9.7 | 0/50 | 0.0 | 6/90 | 6.7 | 1/8 | 12.5 | |
| p = 0.053 | p = 0.355 | p = 1.00 | |||||||
| Electrolyte disorder (pseudo-Bartter syndrome, Salt loss syndrome) | 1/28 | 3.6 | 0/48 | 0.0 | 2/98 | 2.0 | 0/8 | 0.0 | |
| p = 0.36 | p = 0.53 | p = 0.389 | |||||||
| Osteoporosis | 3/25 | 12.0 | 7/36 | 19.4 | 7/69 | 10.1 | 3/6 | 50.0 | |
| p = 0.50 | p = 0.72 | p = 0.068 | |||||||
| Diabetes | 1/31 | 3.2 | 0/50 | 0.0 | 4/100 | 4.0 | 1/9 | 11.1 | |
| p = 0.382 | p = 1.00 | p = 0.403 | |||||||
| Liver disease | All | 4/31 | 12.9 | 3/49 | 6.1 | 29/100 | 29.0 | 2/9 | 22.2 |
| p = 0.421 | p = 0.096 | p = 0.602 | |||||||
| cirrhosis with hypertension | 1 | 25.0 | 0 | 0.0 | 9 | 31.0 | 0 | 0.0 | |
| without cirrhosis | 3 | 75.0 | 3 | 100.0 | 20 | 69.0 | 2 | 100.0 | |
| p = 1.00 | p = 1.00 | p = 1.00 | |||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, N.V.; Kashirskaya, N.Y.; Krasovskiy, S.A.; Amelina, E.L.; Kondratyeva, E.I.; Marakhonov, A.V.; Vasilyeva, T.A.; Voronkova, A.Y.; Sherman, V.D.; Ginter, E.K.; et al. Clinical Presentation of the c.3844T>C (p.Trp1282Arg, W1282R) Variant in Russian Cystic Fibrosis Patients. Genes 2020, 11, 1137. https://doi.org/10.3390/genes11101137
Petrova NV, Kashirskaya NY, Krasovskiy SA, Amelina EL, Kondratyeva EI, Marakhonov AV, Vasilyeva TA, Voronkova AY, Sherman VD, Ginter EK, et al. Clinical Presentation of the c.3844T>C (p.Trp1282Arg, W1282R) Variant in Russian Cystic Fibrosis Patients. Genes. 2020; 11(10):1137. https://doi.org/10.3390/genes11101137
Chicago/Turabian StylePetrova, Nika V., Nataliya Y. Kashirskaya, Stanislav A. Krasovskiy, Elena L. Amelina, Elena I. Kondratyeva, Andrey V. Marakhonov, Tatyana A. Vasilyeva, Anna Y. Voronkova, Victoria D. Sherman, Evgeny K. Ginter, and et al. 2020. "Clinical Presentation of the c.3844T>C (p.Trp1282Arg, W1282R) Variant in Russian Cystic Fibrosis Patients" Genes 11, no. 10: 1137. https://doi.org/10.3390/genes11101137