Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates

Abstract
1. Introduction
2. Materials and Methods
2.1. The Consensify Method
- if ni < m or ni > M,
- then no base call is made.
- else if ni > f,
- then k reads are randomly subsampled
- if f of the subsampled reads agree,
- then a base is called
- else, no base call is made.
- else if ni = f,
- if f reads agree,
- then a base is called
- else,
- no base call is made.
2.2. Test Datasets
2.3. Generation of the Consensify Sequences
2.4. Effect of Consensify on Phylogenetic and Clustering Analysis
2.5. Effect of Consensify on Admixture Tests
3. Results
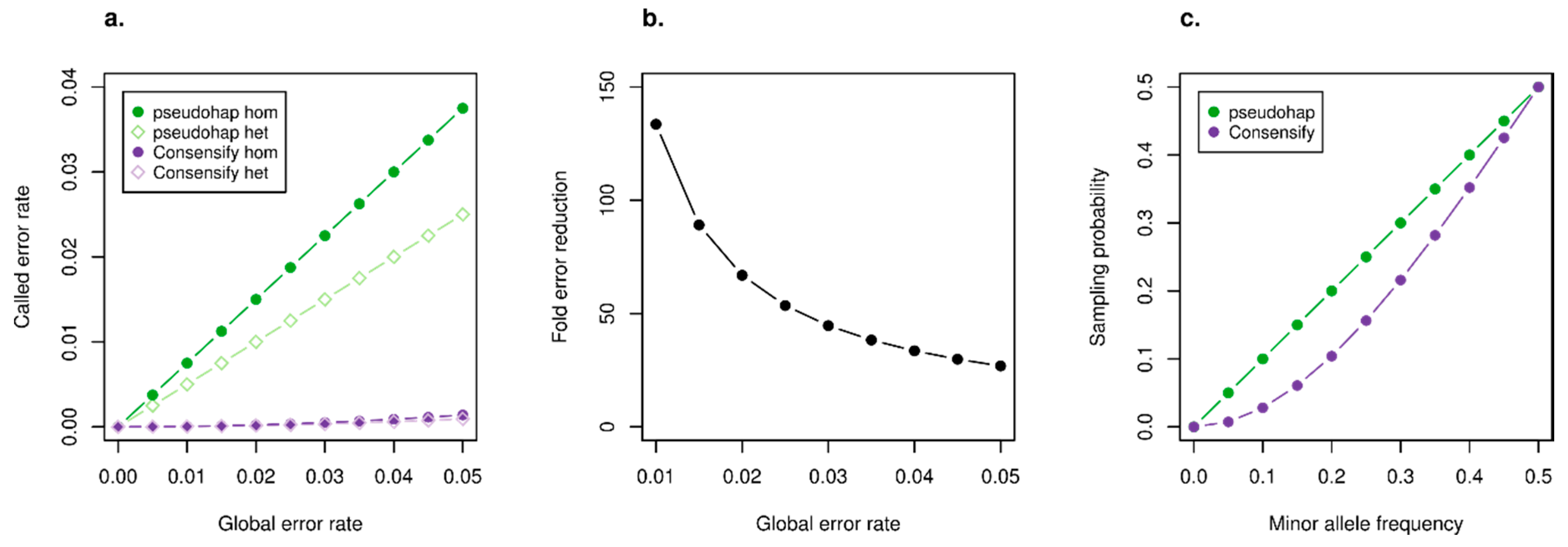
3.1. Statistical Properties of the Consensify Method
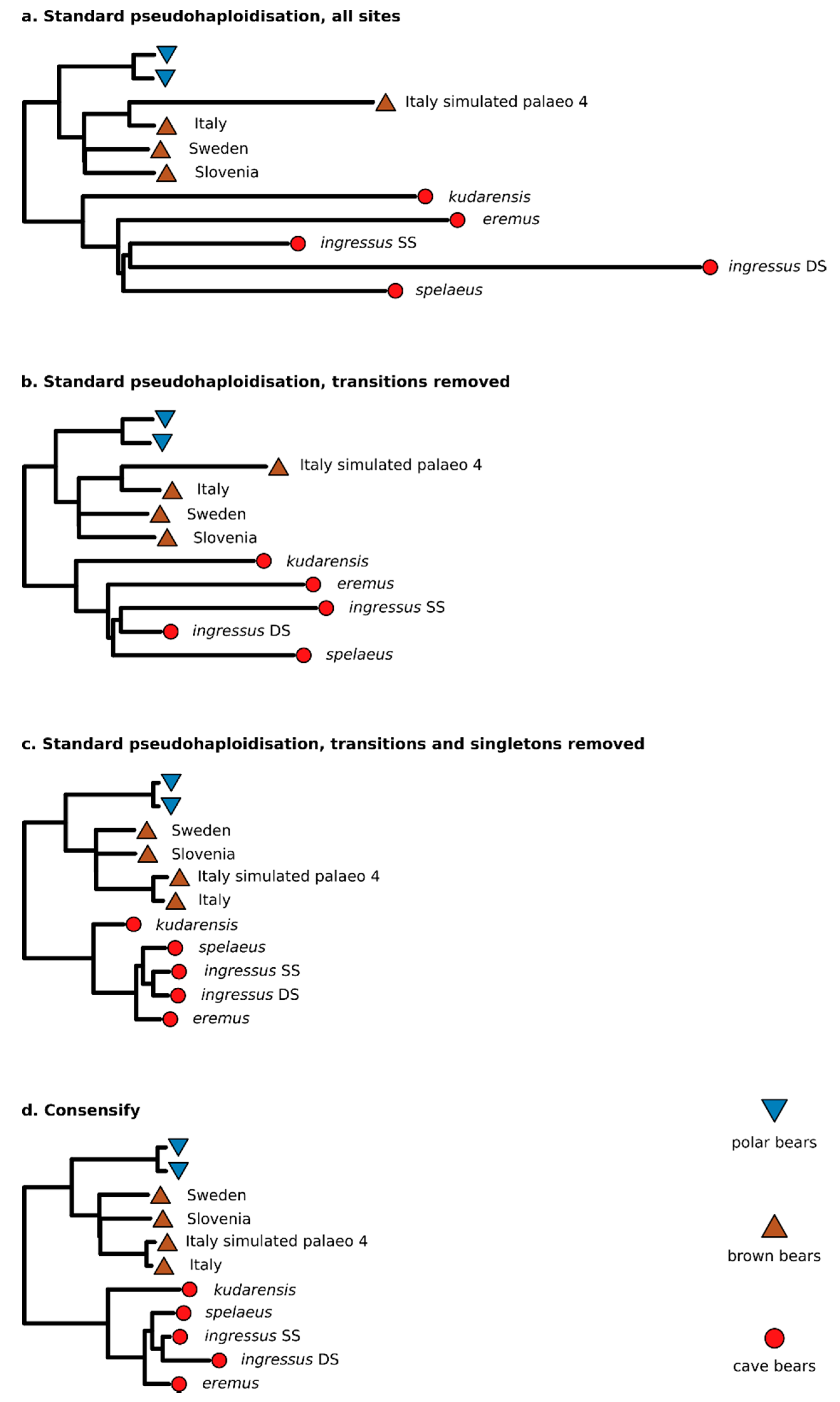
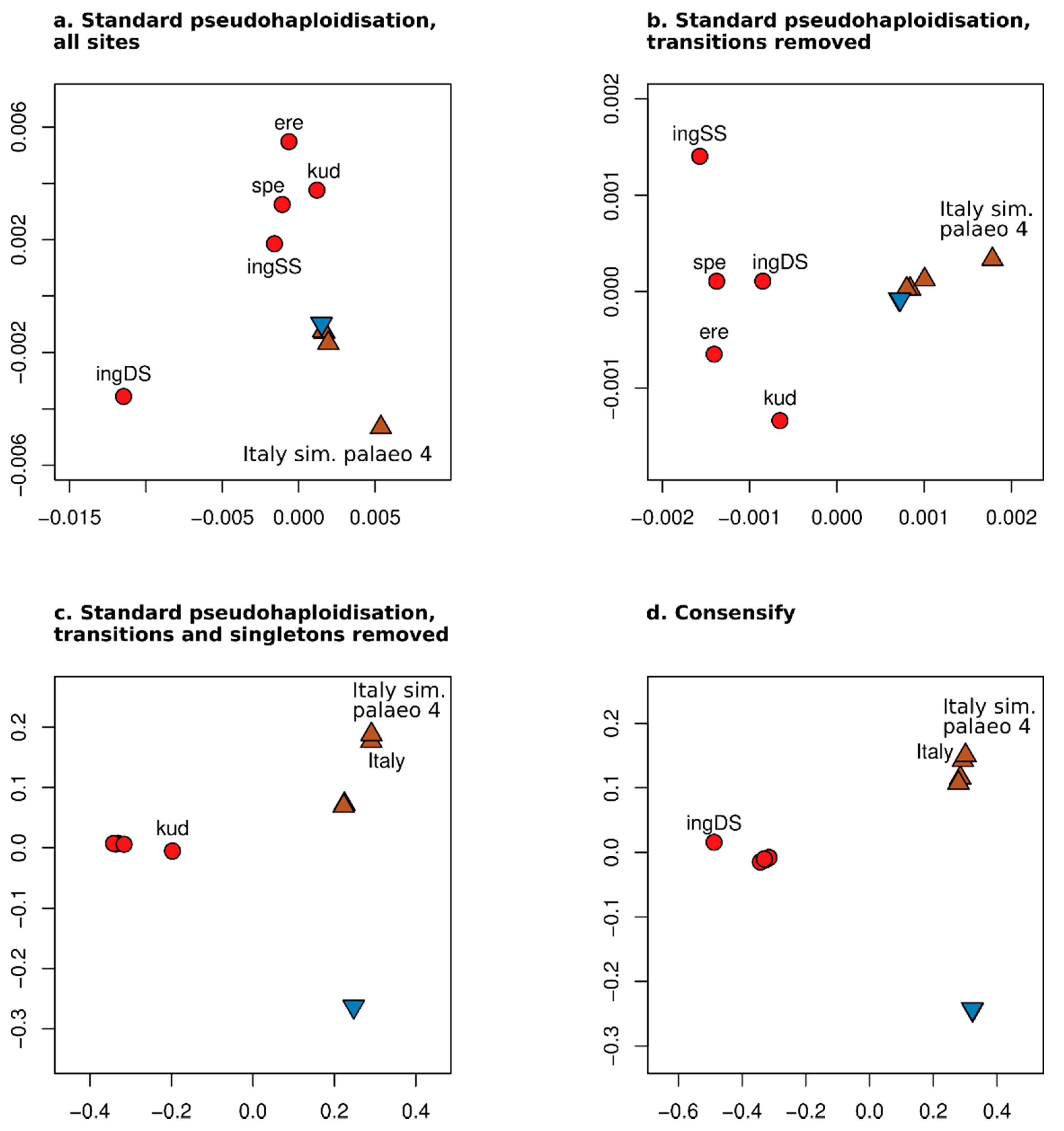
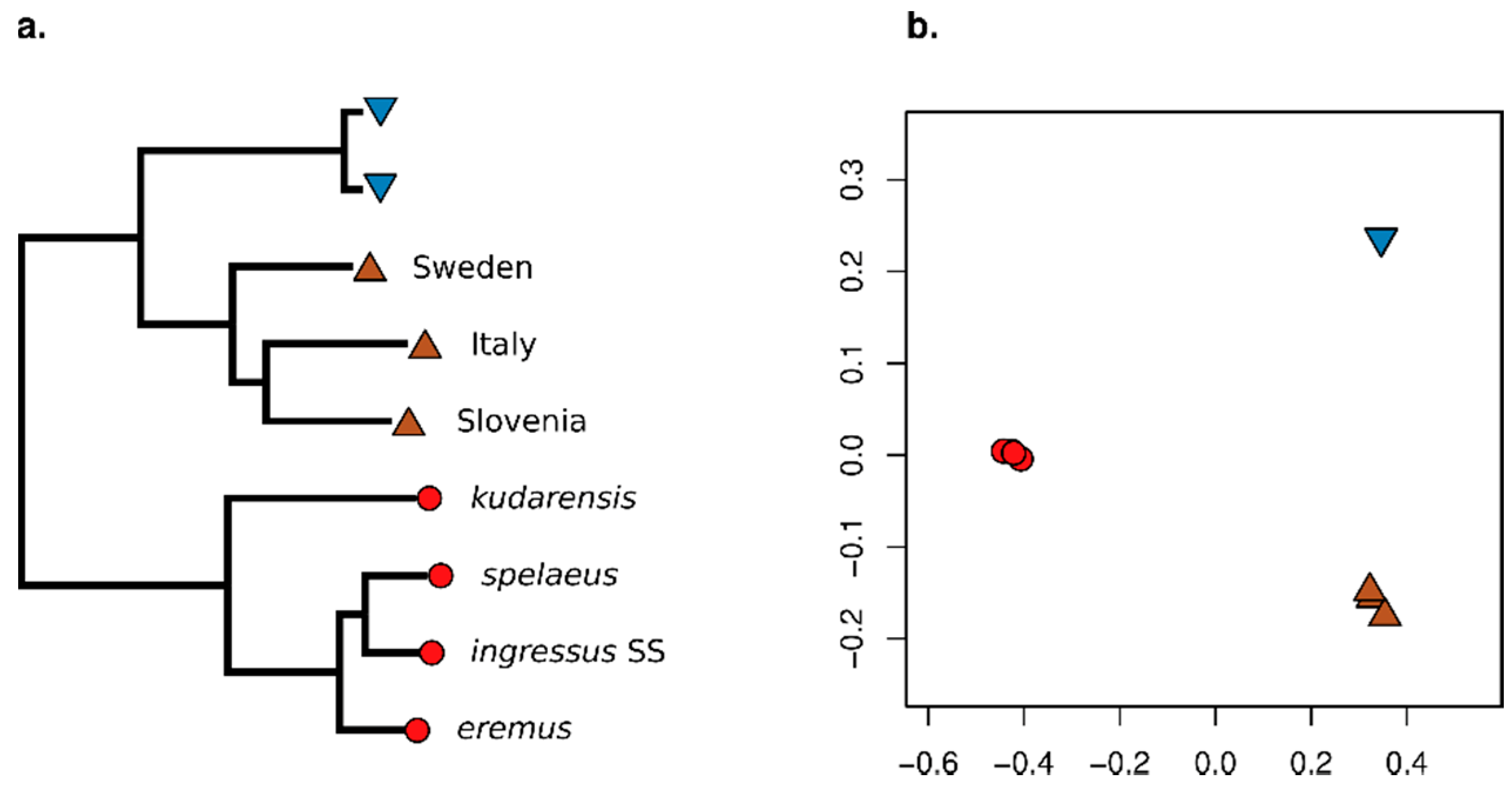
3.2. Effect of Consensify on Phylogenetic and Clustering Analyses
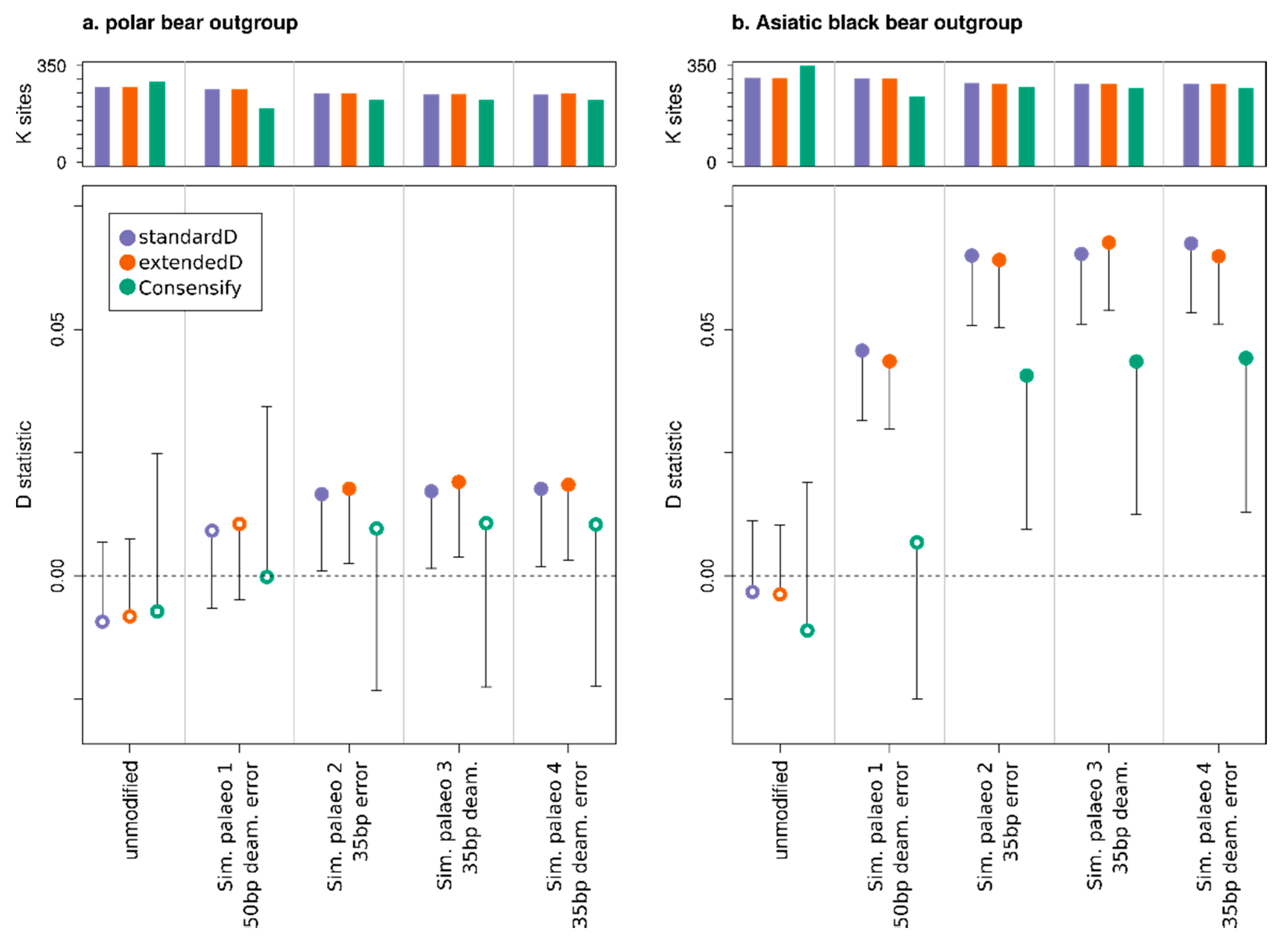
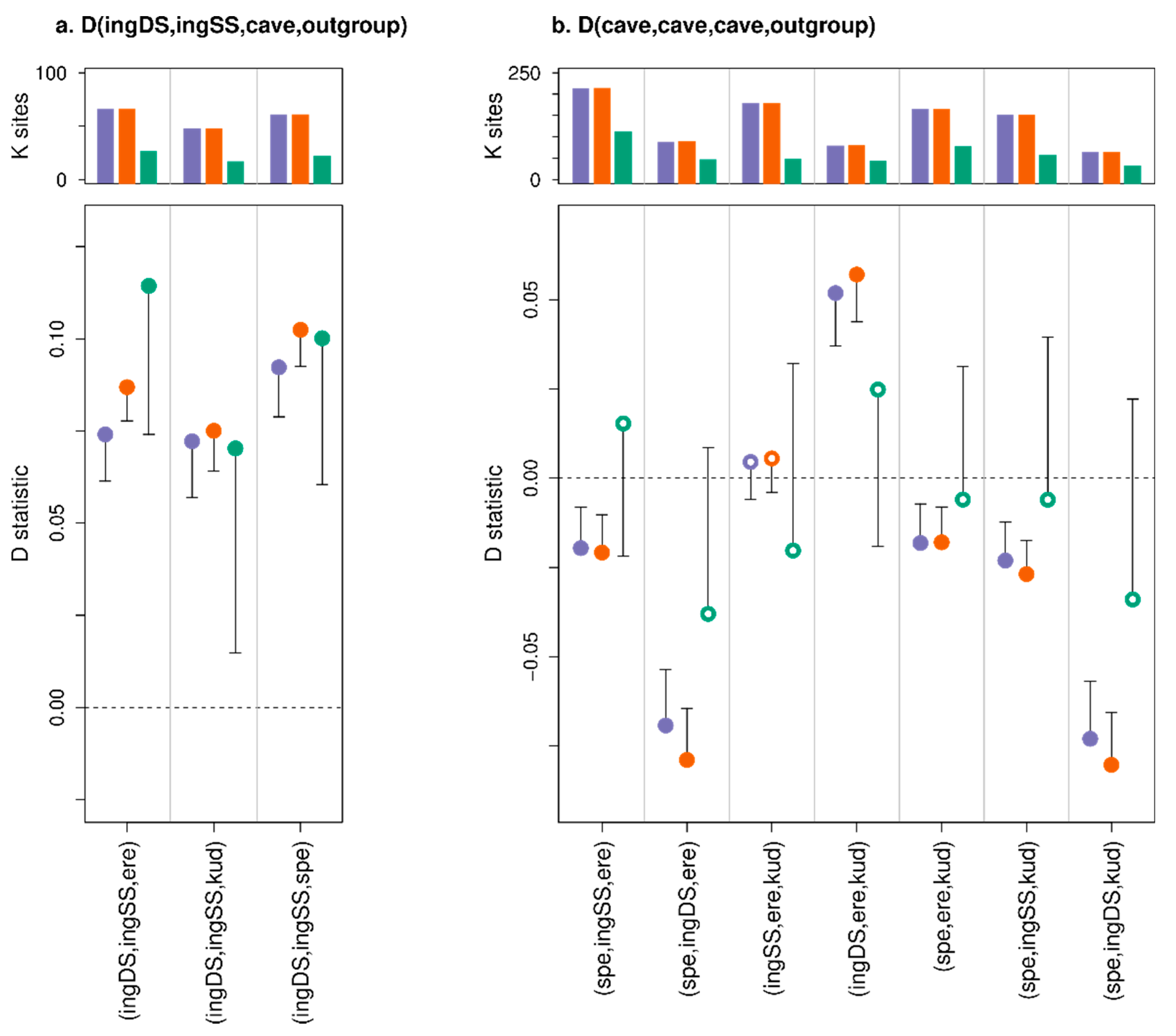
3.3. Effect of Consensify on Admixture Tests
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Briggs, A.W.; Stenzel, U.; Johnson, P.L.F.; Green, R.E.; Kelso, J.; Prüfer, K.; Meyer, M.; Krause, J.; Ronan, M.T.; Lachmann, M.; et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc. Natl. Acad. Sci. USA 2007, 104, 14616–14621. [Google Scholar] [CrossRef] [PubMed]
- Brotherton, P.; Endicott, P.; Sanchez, J.J.; Beaumont, M.; Barnett, R.; Austin, J.; Cooper, A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007, 35, 5717–5728. [Google Scholar] [CrossRef] [PubMed]
- Heyn, P.; Stenzel, U.; Briggs, A.W.; Kircher, M.; Hofreiter, M.; Meyer, M. Road blocks on paleogenomes—Polymerase extension profiling reveals the frequency of blocking lesions in ancient DNA. Nucleic Acids Res. 2010, 38, e161. [Google Scholar] [CrossRef] [PubMed]
- Hofreiter, M.; Jaenicke, V.; Serre, D.; von Haeseler, A.; Pääbo, S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001, 29, 4793–4799. [Google Scholar] [CrossRef]
- Barlow, A.; Cahill, J.A.; Hartmann, S.; Theunert, C.; Xenikoudakis, G.; Fortes, G.G.; Paijmans, J.L.A.; Rabeder, G.; Frischauf, C.; Grandal-d’Anglade, A.; et al. Partial genomic survival of cave bears in living brown bears. Nat. Ecol. Evol. 2018, 2, 1563. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.Y.; et al. A Draft Sequence of the Neandertal Genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef]
- Green, R.E.; Krause, J.; Ptak, S.E.; Briggs, A.W.; Ronan, M.T.; Simons, J.F.; Du, L.; Egholm, M.; Rothberg, J.M.; Paunovic, M.; et al. Analysis of one million base pairs of Neanderthal DNA. Nature 2006, 444, 330–336. [Google Scholar] [CrossRef]
- Orlando, L.; Ginolhac, A.; Zhang, G.; Froese, D.; Albrechtsen, A.; Stiller, M.; Schubert, M.; Cappellini, E.; Petersen, B.; Moltke, I.; et al. Recalibrating Equus evolution using the genome sequence of an early Middle Pleistocene horse. Nature 2013, 499, 74–78. [Google Scholar] [CrossRef]
- Palkopoulou, E.; Lipson, M.; Mallick, S.; Nielsen, S.; Rohland, N.; Baleka, S.; Karpinski, E.; Ivancevic, A.M.; To, T.H.; Kortschak, R.D.; et al. A comprehensive genomic history of extinct and living elephants. Proc. Natl. Acad. Sci. USA 2018, 115, E2566–E2574. [Google Scholar] [CrossRef]
- Skoglund, P.; Ersmark, E.; Palkopoulou, E.; Dalén, L. Ancient Wolf Genome Reveals an Early Divergence of Domestic Dog Ancestors and Admixture into High-Latitude Breeds. Curr. Biol. 2015, 25, 1515–1519. [Google Scholar] [CrossRef]
- Meyer, M.; Kircher, M.; Gansauge, M.T.; Li, H.; Racimo, F.; Mallick, S.; Schraiber, J.G.; Jay, F.; Prüfer, K.; de Filippo, C.; et al. A High-Coverage Genome Sequence from an Archaic Denisovan Individual. Science 2012, 338, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Palkopoulou, E.; Mallick, S.; Skoglund, P.; Enk, J.; Rohland, N.; Li, H.; Omrak, A.; Vartanyan, S.; Poinar, H.; Götherström, A.; et al. Complete Genomes Reveal Signatures of Demographic and Genetic Declines in the Woolly Mammoth. Curr. Biol. 2015, 25, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; de Filippo, C.; Grote, S.; Mafessoni, F.; Korlević, P.; Hajdinjak, M.; Vernot, B.; Skov, L.; Hsieh, P.; Peyrégne, S.; et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 2017, 358, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Kousathanas, A.; Leuenberger, C.; Link, V.; Sell, C.; Burger, J.; Wegmann, D. Inferring Heterozygosity from Ancient and Low Coverage Genomes. Genetics 2017, 205, 317–332. [Google Scholar] [CrossRef]
- Schraiber, J.G. Assessing the Relationship of Ancient and Modern Populations. Genetics 2018, 208, 383–398. [Google Scholar] [CrossRef]
- Hofmanová, Z.; Kreutzer, S.; Hellenthal, G.; Sell, C.; Diekmann, Y.; Díez-del-Molino, D.; van Dorp, L.; López, S.; Kousathanas, A.; Link, V.; et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc. Natl. Acad. Sci. USA 2016, 113, 6886–6891. [Google Scholar] [CrossRef]
- Barlow, A.; Fortes, G.M.G.; Dalen, L.; Pinhasi, R.; Gasparyan, B.; Rabeder, G.; Frischauf, C.; Paijmans, J.L.A.; Hofreiter, M. Massive influence of DNA isolation and library preparation approaches on palaeogenomic sequencing data. BioRxiv 2016, 075911. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M.; Pääbo, S. Ancient DNA Damage. Cold Spring Harb. Perspect. Biol. 2013, 5, a012567. [Google Scholar] [CrossRef]
- Dabney, J.; Meyer, M. Length and GC-biases during sequencing library amplification: A comparison of various polymerase-buffer systems with ancient and modern DNA sequencing libraries. BioTechniques 2012, 52, 87–94. [Google Scholar] [CrossRef]
- Westbury, M.V.; Hartmann, S.; Barlow, A.; Wiesel, I.; Leo, V.; Welch, R.; Parker, D.M.; Sicks, F.; Ludwig, A.; Dalén, L.; et al. Extended and Continuous Decline in Effective Population Size Results in Low Genomic Diversity in the World’s Rarest Hyena Species, the Brown Hyena. Mol. Biol. Evol. 2018, 35, 1225–1237. [Google Scholar] [CrossRef]
- Durand, E.Y.; Patterson, N.; Reich, D.; Slatkin, M. Testing for Ancient Admixture between Closely Related Populations. Mol. Biol. Evol. 2011, 28, 2239–2252. [Google Scholar] [CrossRef] [PubMed]
- Soraggi, S.; Wiuf, C.; Albrechtsen, A. Powerful Inference with the D-Statistic on Low-Coverage Whole-Genome Data. G3: Genes Genomes Genet. 2017, 8, 551–566. [Google Scholar]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. In Mammalian Protein Metabolism; Munro, H.N., Ed.; Academic Press, Inc.: London, UK, 1969; Volume 3, pp. 21–132. [Google Scholar]
- Benazzo, A.; Trucchi, E.; Cahill, J.A.; Delser, P.M.; Mona, S.; Fumagalli, M.; Bunnefeld, L.; Cornetti, L.; Ghirotto, S.; Girardi, M.; et al. Survival and divergence in a small group: The extraordinary genomic history of the endangered Apennine brown bear stragglers. Proc. Natl. Acad. Sci. USA 2017, 114, E9589–E9597. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.A.; Green, R.E.; Fulton, T.L.; Stiller, M.; Jay, F.; Ovsyanikov, N.; Salamzade, R.; St. John, J.; Stirling, I.; Slatkin, M.; et al. Genomic Evidence for Island Population Conversion Resolves Conflicting Theories of Polar Bear Evolution. PLoS Genet. 2013, 9, e1003345. [Google Scholar] [CrossRef] [PubMed]
- Cahill, J.A.; Stirling, I.; Kistler, L.; Salamzade, R.; Ersmark, E.; Fulton, T.L.; Stiller, M.; Green, R.E.; Shapiro, B. Genomic evidence of geographically widespread effect of gene flow from polar bears into brown bears. Mol. Ecol. 2015, 24, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Lammers, F.; Bidon, T.; Pfenninger, M.; Kolter, L.; Nilsson, M.A.; Janke, A. The evolutionary history of bears is characterized by gene flow across species. Sci. Rep. 2017, 7, 46487. [Google Scholar] [CrossRef] [PubMed]
- Gansauge, M.T.; Meyer, M. Single-stranded DNA library preparation for the sequencing of ancient or damaged DNA. Nat. Protoc. 2013, 8, 737–748. [Google Scholar] [CrossRef]
- Henneberger, K.; Barlow, A.; Paijmans, J.L.A. Double-Stranded Library Preparation for Ancient and Other Degraded Samples. In Ancient DNA; Shapiro, B., Barlow, A., Heintzman, P.D., Hofreiter, M., Paijmans, J.L.A., Soares, A.E.R., Eds.; Springer: New York, NY, USA, 2019; Volume 1963, pp. 65–73. ISBN 978-1-4939-9175-4. [Google Scholar]
- Meyer, M.; Kircher, M. Illumina Sequencing Library Preparation for Highly Multiplexed Target Capture and Sequencing. Cold Spring Harb. Protoc. 2010, 2010. [Google Scholar] [CrossRef]
- Basler, N.; Xenikoudakis, G.; Westbury, M.V.; Song, L.; Sheng, G.; Barlow, A. Reduction of the contaminant fraction of DNA obtained from an ancient giant panda bone. BMC Res. Notes 2017, 10, 754. [Google Scholar] [CrossRef]
- Paijmans, J.L.A.; Baleka, S.; Henneberger, K.; Taron, U.H.; Trinks, A.; Westbury, M.V.; Barlow, A. Sequencing single-stranded libraries on the Illumina NextSeq 500 platform. arXiv 2017, arXiv:1711.11004. [Google Scholar]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef]
- Taron, U.H.; Lell, M.; Barlow, A.; Paijmans, J.L.A. Testing of Alignment Parameters for Ancient Samples: Evaluating and Optimizing Mapping Parameters for Ancient Samples Using the TAPAS Tool. Genes 2018, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, Q.; Ma, S.; Ma, T.; Shan, L.; Wang, X.; Nie, Y.; Ning, Z.; Yan, L.; Xiu, Y.; et al. Comparative genomics reveals convergent evolution between the bamboo-eating giant and red pandas. Proc. Natl. Acad. Sci. USA 2017, 114, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Abella, J.; Alba, D.M.; Robles, J.M.; Valenciano, A.; Rotgers, C.; Carmona, R.; Montoya, P.; Morales, J. Kretzoiarctos gen. nov., the Oldest Member of the Giant Panda Clade. PLoS ONE 2012, 7, e48985. [Google Scholar] [CrossRef]
- Krause, J.; Unger, T.; Noçon, A.; Malaspinas, A.S.; Kolokotronis, S.O.; Stiller, M.; Soibelzon, L.; Spriggs, H.; Dear, P.H.; Briggs, A.W.; et al. Mitochondrial genomes reveal an explosive radiation of extinct and extant bears near the Miocene-Pliocene boundary. BMC Evol. Biol. 2008, 8, 220. [Google Scholar] [CrossRef]
- Liu, S.; Lorenzen, E.D.; Fumagalli, M.; Li, B.; Harris, K.; Xiong, Z.; Zhou, L.; Korneliussen, T.S.; Somel, M.; Babbitt, C.; et al. Population Genomics Reveal Recent Speciation and Rapid Evolutionary Adaptation in Polar Bears. Cell 2014, 157, 785–794. [Google Scholar] [CrossRef]
- Günther, T.; Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. BioRxiv 2018, 487983. [Google Scholar] [CrossRef]
- Sheng, G.L.; Basler, N.; Ji, X.P.; Paijmans, J.L.A.; Alberti, F.; Preick, M.; Hartmann, S.; Westbury, M.V.; Yuan, J.X.; Jablonski, N.G.; et al. Paleogenome Reveals Genetic Contribution of Extinct Giant Panda to Extant Populations. Curr. Biol. 2019, 29, 1695–1700.e6. [Google Scholar] [CrossRef] [PubMed]
- Korneliussen, T.S.; Albrechtsen, A.; Nielsen, R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinform. 2014, 15, 356. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Hofreiter, M.; Rabeder, G.; Jaenicke-Després, V.; Withalm, G.; Nagel, D.; Paunovic, M.; Jambresić, G.; Pääbo, S. Evidence for Reproductive Isolation between Cave Bear Populations. Curr. Biol. 2004, 14, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Stiller, M.; Molak, M.; Prost, S.; Rabeder, G.; Baryshnikov, G.; Rosendahl, W.; Münzel, S.; Bocherens, H.; Grandal-d’Anglade, A.; Hilpert, B.; et al. Mitochondrial DNA diversity and evolution of the Pleistocene cave bear complex. Quat. Int. 2014, 339–340, 224–231. [Google Scholar] [CrossRef]
- Pinhasi, R.; Fernandes, D.; Sirak, K.; Novak, M.; Connell, S.; Alpaslan-Roodenberg, S.; Gerritsen, F.; Moiseyev, V.; Gromov, A.; Raczky, P.; et al. Optimal Ancient DNA Yields from the Inner Ear Part of the Human Petrous Bone. PLoS ONE 2015, 10, e0129102. [Google Scholar] [CrossRef]
- Alberti, F.; Gonzalez, J.; Paijmans, J.L.A.; Basler, N.; Preick, M.; Henneberger, K.; Trinks, A.; Rabeder, G.; Conard, N.J.; Münzel, S.C.; et al. Optimized DNA sampling of ancient bones using Computed Tomography scans. Mol. Ecol. Resour. 2018, 18, 1196–1208. [Google Scholar] [CrossRef]
- Damgaard, P.B.; Margaryan, A.; Schroeder, H.; Orlando, L.; Willerslev, E.; Allentoft, M.E. Improving access to endogenous DNA in ancient bones and teeth. Sci. Rep. 2015, 5, 1–12. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| totA | totC | totG | totT |
|---|---|---|---|
| 0 | 0 | 1 | 2 |
| 0 | 0 | 2 | 0 |
| 1 | 0 | 0 | 1 |
| 4 | 0 | 0 | 0 |
| 0 | 4 | 1 | 0 |
| Dataset | Taxon | Data Type | Reference | Mapped Gb 1 | Coverage 2 | Prop > 2 Reads 3 | Consensify Sites 4 |
|---|---|---|---|---|---|---|---|
| E-VD-1838 | cave bear (spelaeus) | ancient single-stranded | [5] | 4.55215 | 1.87465 | 0.48676 | 971,153,181 |
| GS136_ds | cave bear (ingressus) | ancient double-stranded | [5] | 3.72732 | 1.53497 | 0.28367 | 519,642,820 |
| GS136_ss | cave bear (ingressus) | ancient single-stranded | this study | 6.94074 | 2.85831 | 0.64547 | 1,266,005,835 |
| WK01 | cave bear (eremus) | ancient single-stranded | [5] | 6.12884 | 2.52396 | 0.60009 | 1,210,933,302 |
| HV74 | cave bear (kudarensis) | ancient single-stranded | [5] | 3.76075 | 1.54874 | 0.44215 | 869,048,390 |
| 191Y | brown bear (Slovenia) | modern | [5] | 6.99668 | 2.88135 | 0.54327 | 1,088,167,390 |
| SRS779830 | brown bear (Sweden) | modern | [25] | 6.13821 | 2.52782 | 0.53406 | 1,076,186,512 |
| SRR5878360 | brown bear (Italy) | modern | [23] | 17.5122 | 7.21182 | 0.74861 | 1,553,722,573 |
| simulated palaeo 1 (50 bp, deamination, error) | 10.51792 | 4.33146 | 0.74394 | 1,109,680,441 | |||
| simulated palaeo 2 (35 bp, error) | 7.55782 | 3.11244 | 0.6733 | 1,297,732,079 | |||
| simulated palaeo 3 (35 bp, deamination) | 7.42132 | 3.05622 | 0.66679 | 1,293,235,675 | |||
| simulated palaeo 4 (35 bp, deamination, error) | 7.36212 | 3.03185 | 0.66404 | 1,291,668,999 | |||
| SRS412584 | polar bear | modern | [24] | 6.81197 | 2.80528 | 0.55527 | 1,118,483,213 |
| SRS412585 | polar bear | modern | [24] | 6.02194 | 2.47994 | 0.51659 | 1,025,241,632 |
| ERS781634 | Asiatic black bear | modern | [26] | 13.43641 | 5.53334 | 0.72865 | 1,523,577,868 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barlow, A.; Hartmann, S.; Gonzalez, J.; Hofreiter, M.; Paijmans, J.L.A. Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates. Genes 2020, 11, 50. https://doi.org/10.3390/genes11010050
Barlow A, Hartmann S, Gonzalez J, Hofreiter M, Paijmans JLA. Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates. Genes. 2020; 11(1):50. https://doi.org/10.3390/genes11010050
Chicago/Turabian StyleBarlow, Axel, Stefanie Hartmann, Javier Gonzalez, Michael Hofreiter, and Johanna L. A. Paijmans. 2020. "Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates" Genes 11, no. 1: 50. https://doi.org/10.3390/genes11010050
APA StyleBarlow, A., Hartmann, S., Gonzalez, J., Hofreiter, M., & Paijmans, J. L. A. (2020). Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates. Genes, 11(1), 50. https://doi.org/10.3390/genes11010050