SNP Diversity in CD14 Gene Promoter Suggests Adaptation Footprints in Trypanosome Tolerant N’Dama (Bos taurus) but not in Susceptible White Fulani (Bos indicus) Cattle



Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Sampling
2.2. Genomic DNA Extraction, PCR, and DNA Sequencing
2.3. Sequence Processing and Alignment
2.4. Variant Analysis, Prediction of Transcription Factor Binding Sites, and miRNAs
2.5. Functional Analysis of miRNAs at the Core Promoter of CD14 Gene
2.6. Validation of Selected CD14 Gene Polymorphism by Taqman SNP Genotyping Assay
2.7. Statistics
3. Results
3.1. Sequence Quality Control and Alignments
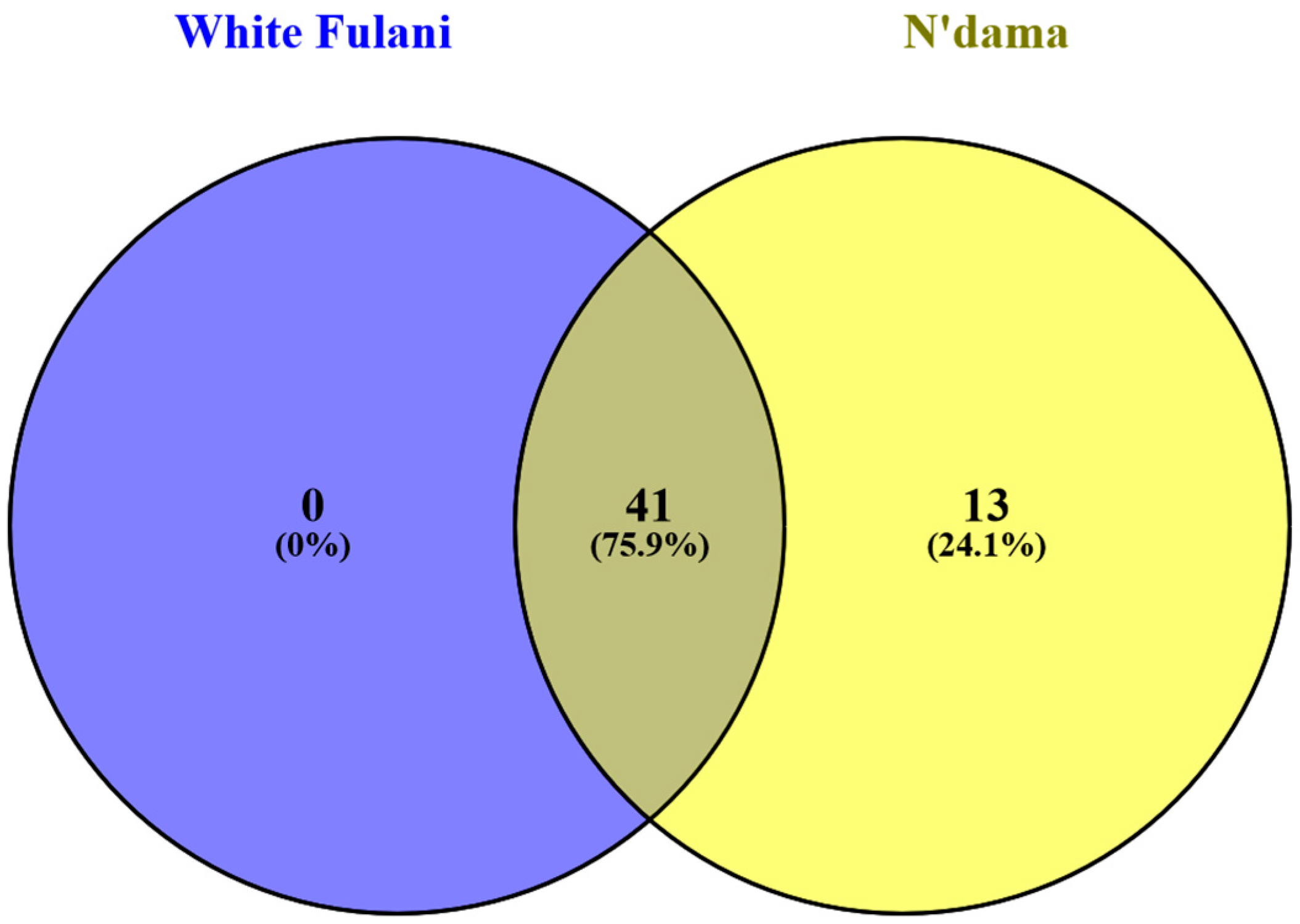
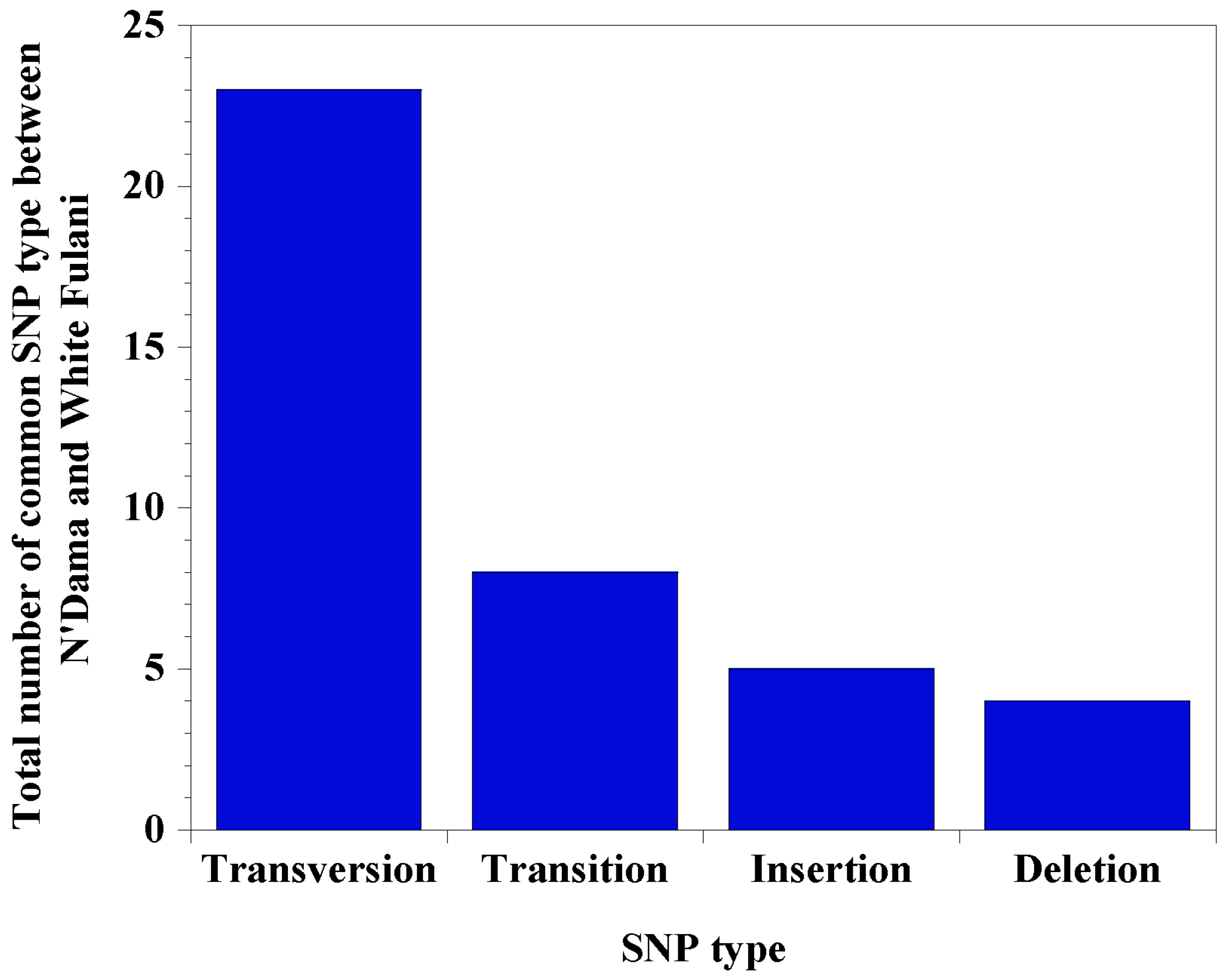
3.2. Nucleotide Mapping, SNP Identification, and Classification
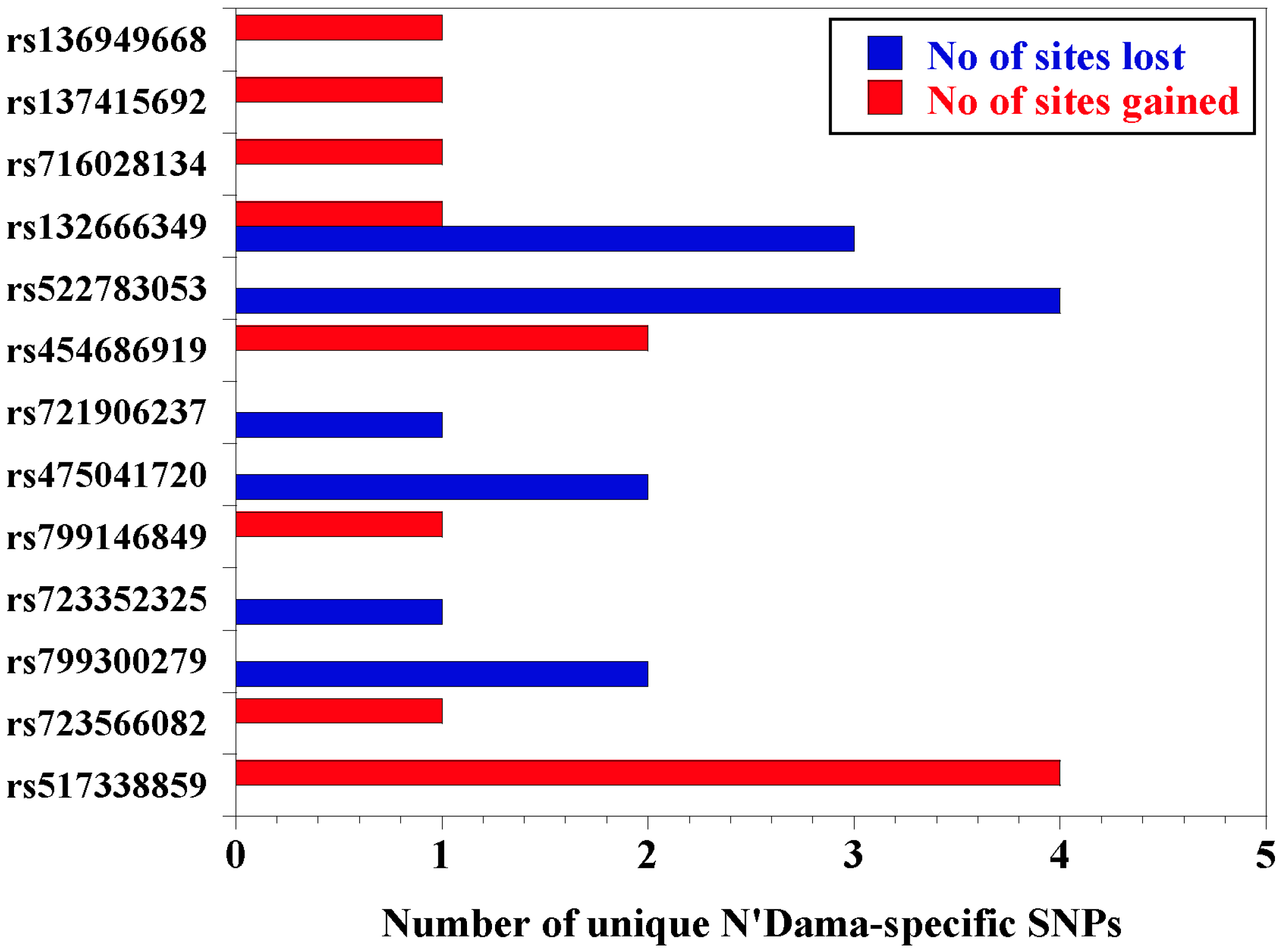
3.3. Prediction of SNP Effects, Nucleotide Diversity, and Transcription Factor Binding Sites
3.4. miRNAs Identification and Functional Classification
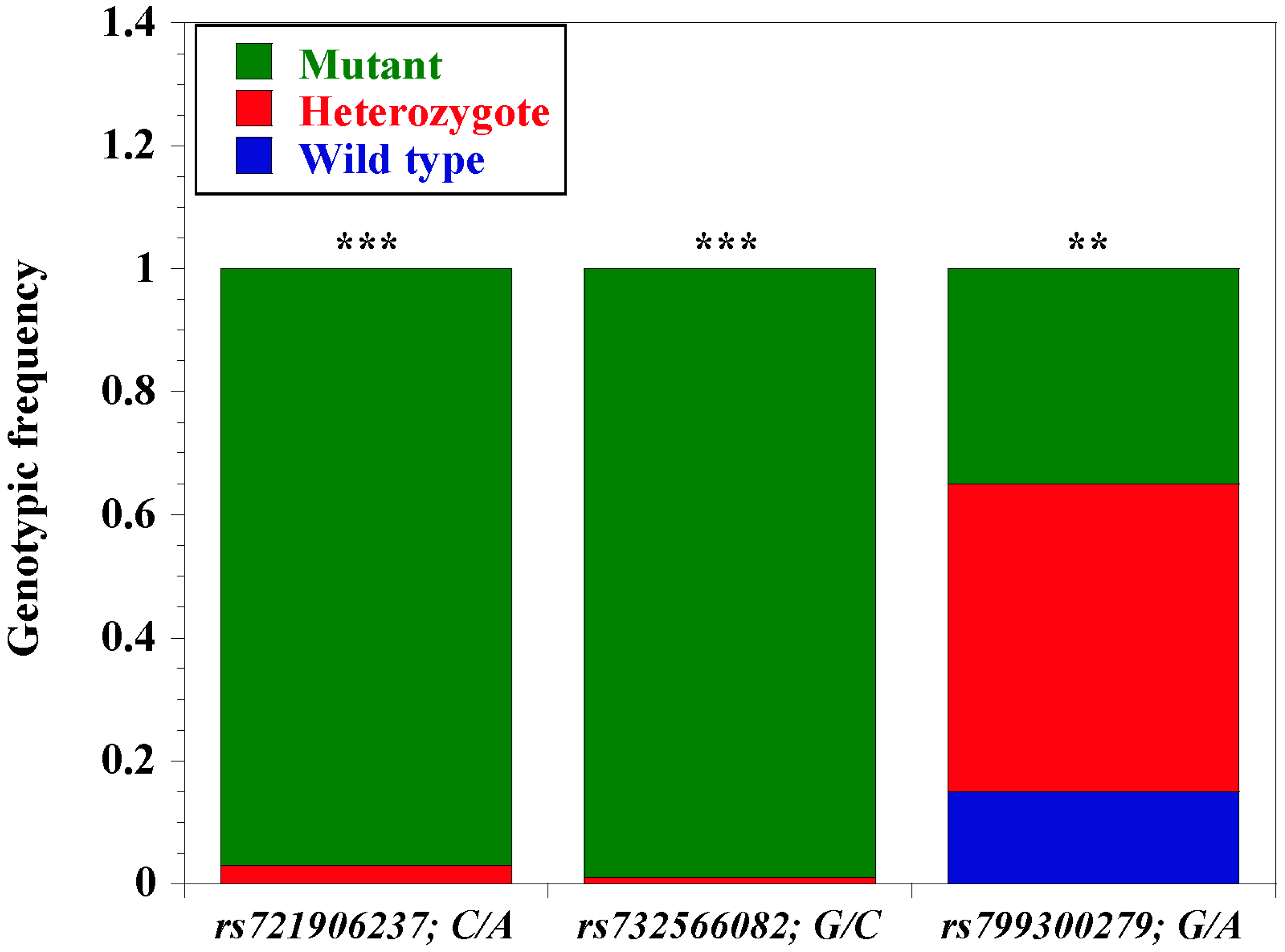
3.5. Allelic, Genotypic, and Haplotype Estimates of Three Loci in N’Dama CD14 Promoter
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bai, Y.; Sartor, M.; Cavalcoli, J. Current status and future perspectives for sequencing livestock genomes. J. Anim. Sci. Biotechnol. 2012, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Goddard, M.E. Uses of genomics in livestock agriculture. Anim. Prod. Sci. 2012, 52, 73–77. [Google Scholar] [CrossRef]
- Zwane, A.A.; Schnabel, R.D.; Hoff, J.; Choudhury, A.; Makgahlela, M.L.; Maiwashe, A.; Van Marle-Koster, E.; Taylor, J.F. Genome-Wide SNP Discovery in Indigenous Cattle Breeds of South Africa. Front. Genet. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Ka, S.; Ha, J.W.; Kim, J.; Yoo, D.; Kim, K.; Lee, H.K.; Lim, D.; Cho, S.; Hanotte, O.; et al. Cattle genome-wide analysis reveals genetic signatures in trypanotolerant N’Dama. BMC Genom. 2017, 18, 371. [Google Scholar] [CrossRef]
- Smetko, A.; Soudre, A.; Silbermayr, K.; Müller, S.; Brem, G.; Hanotte, O.; Boettcher, P.J.; Stella, A.; Mészáros, G.; Wurzinger, M.; et al. Trypanosomosis: Potential driver of selection in African cattle. Front. Genet 2015, 6, 137. [Google Scholar] [CrossRef]
- Tijjani, A.; Utsunomiya, Y.T.; Ezekwe, A.G.; Nashiru, O.; Hanotte, O. Genome Sequence Analysis Reveals Selection Signatures in Endangered Trypanotolerant West African Muturu Cattle. Front. Genet 2019, 10, 442. [Google Scholar] [CrossRef]
- Lemecha, H.; Mulatu, W.; Hussein, I.; Rege, E.; Tekle, T.; Abdicho, S.; Ayalew, W. Response of four indigenous cattle breeds tsetse to natural Ethiopia trypanosomosis challenge in the Ghibe valley of of Ethiopia. Vet. Parasitol. 2006, 141, 165–176. [Google Scholar] [CrossRef]
- Noyes, H.; Brass, A.; Obara, I.; Anderson, S.; Archibald, A.L.; Bradley, D.G.; Fisher, P.; Freeman, A.; Gibson, J.; Gicheru, M.; et al. Genetic and expression analysis of cattle identifies candidate genes in pathways responding to Trypanosoma congolense infection. Proc. Natl. Acad. Sci. USA 2011, 108, 22. [Google Scholar] [CrossRef]
- Auty, H.; Torr, S.J.; Michoel, T.; Jayaraman, S.; Morrison, L.J. Cattle trypanosomosis: The diversity of trypanosomes and implications for disease epidemiology and control. Rev. Sci. Tech. Off. Int. Epiz. 2015, 34, 587–598. [Google Scholar] [CrossRef]
- Zanoni, I.; Granucci, F. Role of CD14 in host protection against infections and in metabolism regulation. Front. Cell. Infect. Microbiol. 2013, 3, 32. [Google Scholar] [CrossRef]
- Baumann, C.L.; Aspalter, I.M.; Sharif, O.; Pichlmair, A.; Bluml, S.; Grebien, F.; Bruckner, M.; Pasierbek, P.; Aumayr, K.; Planyavsky, M.; et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J. Exp. Med. 2010, 207, 2689–2701. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, T.A.; Zorzetto-Fernandes, A.L.V.; Cecílio, N.T.; Sardinha-Silva, A.; Fernandes, F.F.; Roque-Barreira, M.C. CD14 is critical for TLR2-mediated M1 macrophage activation triggered by N-glycan recognition. Sci. Rep. 2017, 7, 7083. [Google Scholar] [CrossRef] [PubMed]
- Ibeagha-Awemu, E.M.; Lee, J.W.; Ibeagha, A.E.; Zhao, X. Bovine CD14 gene characterization and relationship between polymorphisms and surface expression on monocytes and polymorphonuclear neutrophils. BMC Genet. 2008, 9, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Selvan, A.S.; Gupta, I.D.; Verma, A.; Chaudhari, M.V.; Magotra, A. Molecular characterization and combined genotype association study of bovine cluster of differentiation 14 gene with clinical mastitis in crossbred dairy cattle. Vet World 2016, 9, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Gao, W.N.; Chen, F.; Ma, B.B.; Zhou, F.; Hu, Z.G.; Long, T.; Zhao, Z.Q. CD14 gene polymorphisms associated with increased risk of bovine tuberculosis in Chinese Holstein cows. Vet. J. 2018, 232, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Oakley, M.S.; Majam, V.; Mahajan, B.; Gerald, N.; Anantharaman, V.; Ward, J.M.; Faucette, L.J.; McCutchan, T.F.; Zheng, H.; Terabe, M.; et al. Pathogenic roles of CD14, galectin-3 and OX40 during experimental cerebral malaria in mice. PLoS ONE 2009, 4, e6793. [Google Scholar] [CrossRef][Green Version]
- Liu, H.H.; Hu, Y.; Zheng, M.; Suhoski, M.M.; Engleman, E.G.; Dill, D.L.; Hudnall, M.; Wang, J.; Spolski, R.; Leonard, W.J.; et al. Cd14 SNPs regulate the innate immune response. Mol. Immunol. 2012, 51, 112–127. [Google Scholar] [CrossRef][Green Version]
- Ojurongbe, O.; Funwei, R.I.; Snyder, T.; Aziz, N.; Li, Y.; Falade, C.; Thomas, B.N. Genetic diversity of CD14 promoter gene polymorphism (rs2569190) is associated with regulation of parasitemia but not susceptibility to Plasmodium falciparum infection. Infect. Dis. 2017, 10, 1–6. [Google Scholar] [CrossRef]
- Ashley, K.; Harrison, H.; Chan, P.H.; Sothoeun, S.; Young, J.R.; Windsor, P.A.; Bush, R.D. Livestock and livelihoods of smallholder cattle-owning households in Cambodia: The contribution of on-farm and off-farm activities to income and food security. Trop. Anim. Health Prod. 2018, 50, 1747–1761. [Google Scholar] [CrossRef]
- Vercelli, D.; Baldini, M.; Martinez, F.D. The monocyte/IgE connection: May polymorphisms in the CD14 gene teach us about IgE regulation? Int. Arch. Allergy Immunol. 2001, 124, 20–24. [Google Scholar] [CrossRef]
- Baldini, M.; Lohman, I.C.; Halonen, M.; Erickson, R.P.; Holt, P.G.; Martinez, F.D. A polymorphism in the 5’flanking region of the CD14 gene is associated with circulating soluble CD14 levels and with total serum immunoglobulin E. Am. J. Respir. Cell. Mil. Biol. 1999, 20, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.N.; Diallo, D.A.; Moulds, J.M. Circulating immune complex levels are associated with disease severity and seasonality in children with malaria from Mali. Biomark. Insights. 2012, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.N.; Petrella, C.R.; Thakur, T.J.; Crespo, S.R.; Diallo, D.A. Genetic polymorphism of Plasmodium falciparum merozoite surface protein-1 and 2 and diversity of drug resistance genes in blood donors from Bamako, Mali. Infect. Dis. 2012, 6, 49–57. [Google Scholar] [CrossRef]
- Duru, K.C.; Thomas, B.N. Stability and genetic diversity of glutamate-rich protein (GLURP) in Plasmodium falciparum isolates from sub-Saharan Africa. Micro. Insig. 2014, 7, 35–39. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, F.; Chen, S.; Wan, J.; Wang, G. Methods of MicroRNA promoter prediction and transcription factor mediated regulatory network. BioMed Res. Int. 2017. [Google Scholar] [CrossRef]
- Morenikeji, O.B.; Hawkes, M.E.; Hudson, A.O.; Thomas, B.N. Computational Network Analysis Identifies Evolutionarily Conserved miRNA Gene Interactions Potentially Regulating Immune Response in Bovine Trypanosomosis. Front. Microbiol. 2019, 10, 2010. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk--database: Prediction of possible miRNA binding sites by walking the genes of three genomes. J Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. Mirwalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. Mirbase: Anointing high confidence microRNAs using deep sequencing data. Nucelic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Zagganas, K.; Paraskevopoulou, M.D.; Georgakilas, G.; Karagkouni, D.; Vergoulis, T.; Dalamagas, T.; Hatzigeorgiou, A.G. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fan, M.; Zhang, X.; Huang, F.; Wu, K.; Zhang, J.; Liu, J.; Huang, Z.; Luo, H.; Tao, L.; et al. Cellular microRNAs up-regulate transcription via interaction with promoter TATA-box motifs. RNA 2014, 20, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Solé, X.; Guinó, E.; Valls, J.; Iniesta, R.; Moreno, V. SNPStats: A web tool for the analysis of association studies. Bioinformatics 2006, 22, 1928–1929. [Google Scholar] [CrossRef]
- Bahbahani, H.; Tijjani, A.; Mukasa, C.; Wragg, D.; Almathen, F.; Nash, O.; Akpa, G.N.; Mbole-Kariuki, M.; Malla, S.; Woolhouse, M.; et al. Signatures of selection for environmental adaptation and zebu × taurine hybrid fitness in east african shorthorn zebu. Front. Genet. 2017, 8, 68. [Google Scholar] [CrossRef]
- Sinnett, D.; Beaulieu, P.; Bélanger, H.; Lefebvre, J.F.; Langlois, S.; Théberge, M.C.; Drouin, S.; Zotti, C.; Hudson, T.J.; Labuda, D. Detection and characterization of DNA variants in the promoter regions of hundreds of human disease candidate genes. Genomics 2006, 87, 704–710. [Google Scholar] [CrossRef]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef]
- Beecher, C.; Daly, M.; Childs, S.; Berry, D.P.; Magee, D.A.; McCarthy, T.V.; Giblin, L. Polymorphisms in bovine immune genes and their associations with somatic cell count and milk production in dairy cattle. BMC Genet. 2010, 11, 99. [Google Scholar] [CrossRef]
- Pal, A.; Sharma, A.; Bhattacharya, T.K.; Chatterjee, P.N.; Chakravarty, A.K. Molecular characterization and SNP detection of CD14 gene of crossbred cattle. Mol. Bio. Inter. 2011, 507, 346. [Google Scholar] [CrossRef]
- Morenikeji, O.B.; Thomas, B.N. Disease tolerance and immune response in bovine trypanosomiasis is mediated by CD14 promoter gene. J. Immunol. 2019, 73, 17. [Google Scholar]
- Lyons, D.M.; Lauring, A.S. Evidence for the selective basis of transition-to-transversion substitution bias in two RNA viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef] [PubMed]
- Doud, M.B.; Bloom, J.D. Accurate measurement of the effects of all amino-acid mutations on influenza hemagglutinin. Viruses 2016, 8, 155. [Google Scholar] [CrossRef]
- Haddox, H.K.; Dingens, A.S.; Bloom, J.D. Experimental estimation of the effects of all amino-acid mutations to HIV’s envelope protein on viral replication in cell culture. PLoS Pathog. 2016, 12, e1006114. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, Y.X.; Hewett-Emmett, D.; Boerwinkle, E. Investigating single nucleotide polymorphism (SNP) density in the human genome and its implications for molecular evolution. Gene 2003, 312, 207–213. [Google Scholar] [CrossRef]
- Morenikeji, O.B.; Thomas, B.N. In silico analyses of CD14 molecule reveal significant evolutionary diversity, potentially associated with speciation and variable immune response in mammals. PeerJ 2019, 7, e7325. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhao, Z.Q.; Chen, F.; Zhang, L.; Li, G.D.; Ma, K.W.; Bai, X.F.; Zuo, Y.J. Polymorphisms in the promoter of the CD14 gene and their associations with susceptibility to pulmonary tuberculosis. Tissue Antigens 2012, 80, 437–443. [Google Scholar] [CrossRef]
- Yuan, Q.; Chen, H.; Zheng, X.; Chen, X.; Li, Q.; Zhang, Y.; Shi, T.; Zhou, J.; Chen, Q.; Yu, S. The association between C-159T polymorphism in CD14 gene and susceptibility to tuberculosis: A meta-analysis. Mol. Biol. Rep. 2014, 41, 7623–7629. [Google Scholar] [CrossRef] [PubMed]
- Lomelin, D.; Jorgenson, E.; Risch, N. Human genetic variation recognizes functional elements in noncoding sequence. Genome Res. 2009, 20, 311–319.33. [Google Scholar] [CrossRef]
- Jim, J.L.; Shaun, E.G.; Shannon, M.H.; Elisabeth, A.G.; Qin, W. Characterization of cis-Regulatory Elements and Transcription Factor Binding: Gel Mobility Shift Assay. Method. Mol. Biol. 2007, 366, 183–201. [Google Scholar] [CrossRef]
- Natalia, J.; Martinez, A.; Albertha, J.M.W. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 2009, 31, 435–445. [Google Scholar] [CrossRef]
- Tomso, D.J.; Bell, D.A. Sequence context at human single nucleotide polymorphisms: Overrepresentation of CpG dinucleotide at polymorphic sites and suppression of variation in CpG islands. J. Mol. Biol. 2003, 327, 303–308. [Google Scholar] [CrossRef]
- Adefenwa, M.A.; Agaviezor, B.O.; Peters, S.O.; Wheto, M.; Ekundayo, O.J.; Okpeku, M.; Oboh, B.O.; Adekoya, K.O.; Ikeobi, C.O.N.; De Donato, M.; et al. Novel intron 2 polymorphism in the melanophilin gene is in Hardy-Weinberg equilibrium and not associated with coat color in goats. Open J. Genet. 2013, 3, 195–200. [Google Scholar] [CrossRef]
- Chuang, J.H.; Li, H. Functional bias and spatial organization of genes in mutational hot and cold regions in the human genome. PLoS Biol. 2004, 10, 1371. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Taylor, J.F.; Van Tassell, C.P.; Barendse, W.; Eversole, K.A.; Gill, C.A.; Gill, C.A.; Green, R.D.; Hamernik, D.L.; Kappes, S.M.; et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 2009, 324, 528–532. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Schröder, N.W.; Opitz, B.; Lamping, N.; Michelsen, K.S.; Zähringer, U.; Göbel, U.B.; Schumann, R.R. Involvement of lipopolysaccharide binding protein, CD14, and Toll-like receptors in the initiation of innate immune responses by Treponema glycolipids. J. Immunol. 2000, 165, 2683–2693. [Google Scholar] [CrossRef]
- Karki, R.; Pandya, D.; Elston, R.C.; Ferlini, C. Defining mutation and polymorphism in the era of personal genomics. BMC Med. Genom. 2015, 8, 37. [Google Scholar] [CrossRef]
- Huang, J.; Liu, L.; Wang, H.; Zhang, C.; Ju, Z.; Wang, C.; Zhong, J. Variants and gene expression of the TLR2 gene and susceptibility to mastitis in cattle. Asian J. Anim. Vet. Adv. 2011, 6, 51–61. [Google Scholar] [CrossRef][Green Version]
- Zhang, Z.; Wang, X.; Li, R.; Ju, Z.; Qi, C.; Zhang, Y.; Guo, F.; Luo, G.; Li, Q.; Wang, C.; et al. Genetic mutations potentially cause two novel NCF1 splice variants upregulated in the mammary gland, blood and neutrophil of cows infected by Escherichia coli. Microbiol. Res. 2015, 174, 24–32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S/N | SNP ID | Position | Mutation | Substitution Type | Consequence on Transcription Sites | |
|---|---|---|---|---|---|---|
| Number of Sites Lost | Number of Sites Gained | |||||
| 1 | rs471069687 | 53449742 | C → A | Transversion | 2 | 3 |
| 2 | rs438450850 | 53449670 | C → T | Transversion | 1 | 0 |
| 3 | rs472947228 | 53449666 | C → A | Transversion | 0 | 1 |
| 4 | rs452987322 | 53449661 | G → C | Transversion | 3 | 0 |
| 5 | rs517148865 | 53449741 | G → C | Transversion | 1 | 1 |
| 6 | rs442454268 | 53449654 | G → A | Transition | 2 | 2 |
| 7 | rs522987651 | 51766435 | -/C | Insertion | 0 | 1 |
| 8 | rs520985067 | 51766431 | T/- | Deletion | 1 | 0 |
| 9 | rs473919411 | 53449641 | T → A | Transversion | 2 | 4 |
| 10 | rs455092482 | 53449635 | A → T | Transversion | 4 | 2 |
| 11 | rs438139354 | 53449628 | A → C | Transversion | 4 | 2 |
| 12 | rs469547425 | 53449627 | G → C | Transversion | 4 | 1 |
| 13 | rs452652871 | 53449622 | T → C | Transition | 1 | 2 |
| 14 | rs515988451 | 51766408 | -/G | Insertion | - | - |
| 15 | rs523833822 | 51766400 | T/- | Deletion | - | - |
| 16 | rs518926579 | 51766395 | -/G | Insertion | - | - |
| 17 | rs526768201 | 51766392 | T/- | Deletion | - | - |
| 18 | rs452331337 | 53449596 | G → T | Transversion | 1 | 0 |
| 19 | rs443071218 | 51766373 | (T)12/(T)13/(T)14 | Tandem repeat | - | - |
| 20 | rs439484127 | 51766372 | -/T | Insertion | - | - |
| 21 | rs730878557 | 51766372 | T/- | Deletion | - | - |
| 22 | rs440282053 | 53449583 | A → T | Transversion | 1 | 7 |
| 23 | rs797368076 | 51766324 | -/T | Insertion | - | - |
| 24 | rs210109064 | 53449421 | G → A | Transition | 2 | 1 |
| 25 | rs473185091 | 53449334 | C → T | Transition | 1 | 1 |
| 26 | rs521360374 | 53449277 | C → T | Transition | 1 | 2 |
| 27 | rs454858160 | 53449264 | G → T | Transversion | 0 | 1 |
| 28 | rs432371570 | 53449221 | C → A | Transversion | 1 | 3 |
| 29 | rs466880145 | 53449214 | A → C | Transversion | 3 | 2 |
| 30 | rs446947915 | 53449212 | T → C | Transition | 3 | 3 |
| 31 | rs481617328 | 53449165 | T → G | Transversion | 1 | 1 |
| 32 | rs467889921 | 53449162 | G → T | Transversion | 1 | 4 |
| 33 | rs450747566 | 53449152 | G → T | Transversion | 0 | 11 |
| 34 | rs482000686 | 53449147 | G → C | Transversion | 5 | 9 |
| 35 | rs458853723 | 53449144 | T → G | Transversion | 5 | 2 |
| 36 | rs438670802 | 53449139 | G → T | Transversion | 5 | 1 |
| 37 | rs479468105 | 53449129 | A → T | Transversion | 1 | 0 |
| 38 | rs459318293 | 53449109 | T → C | Transition | 2 | 7 |
| 39 | rs442402639 | 53449098 | A → C | Transversion | 1 | 6 |
| 40 | rs473930339 | 53449097 | C → A | Transversion | 5 | 2 |
| 41 | rs456854916 | 53449093 | T → C | Transition | 5 | 12 |
| Total: 69 | Total: 89 | |||||
| S/No | SNP ID | Position | Mutation | Substitution Type | Consequences on Transcription Sites | |
|---|---|---|---|---|---|---|
| Number of Sites Lost | Number of Sites Gained | |||||
| 1 | rs517338859 | 53449994 | G → A | Transition | 0 | 4 |
| 2 | rs723566082 | 51766726 | G → C | Transversion | 0 | 1 |
| 3 | rs799300279 | 51766718 | A → G | Transition | 2 | 0 |
| 4 | rs723352325 | 51766710 | C → T | Transversion | 1 | 0 |
| 5 | rs799146849 | 51766708 | A → G | Transition | 0 | 1 |
| 6 | rs475041720 | 51766680 | G → C | Transversion | 2 | 0 |
| 7 | rs721906237 | 51766666 | C → A | Transversion | 1 | 0 |
| 8 | rs454686919 | 51766632 | C → A | Transversion | 0 | 2 |
| 9 | rs522783053 | 51766588 | G → T | Transversion | 4 | 0 |
| 10 | rs132666349 | 53449790 | C → T | Transition | 3 | 1 |
| 11 | rs716028134 | 53449782 | T → G | Transversion | 0 | 1 |
| 12 | rs137415692 | 53449769 | A → C | Transversion | 0 | 1 |
| 13 | rs136949668 | 51766556 | A/- | Deletion | 0 | 1 |
| Total: 13 | Total: 12 | |||||
| TFBS | TFBS Information | Position | Strand | Matrix Simulation | Sequence |
|---|---|---|---|---|---|
| IRF2.01 | Interferon regulatory factor 2 | 122 | (−) | 0.945 | caatgaatgaaagtGAAAagtgaaa |
| ZNF219.0 | Kruppel-like zinc finger protein 219 | 213 | (−) | 0.920 | caggcCCCCcttccctgggattc |
| GCM1.03 | Glial cells missing homolog 1 (secondary DNA binding preference) | 222 | (−) | 0.851 | gtgacCCCCcataga |
| PLAGL1.02 | Pleiomorphic adenoma gene-like 1 (secondary DNA binding preference) | 225 | (+) | 0.816 | atggGGGGtcacacagagtcaga |
| KLF12.01 | Krueppel-like factor 12 (AP-2rep) | 338 | (+) | 0.938 | ggtcccaGTGGttaagact |
| HMBOX.01 | Homeobox containing 1 | 340 | (+) | 0.834 | tcccagtgGTTAagact |
| ZTRE.01 | Zinc transcriptional regulatory element | 358 | (−) | 0.963 | gtgGGAGtgcagtctta |
| E2F.02 | E2F, involved in cell cycle regulation, interacts with Rb p107 protein | 420 | (+) | 0.849 | cagcagggcCAAAaaaa |
| OVOL1.01 | Zinc finger transcription factor OVO homolog-like 1 | 438 | (+) | 0.891 | aaatctGTTActttc |
| CP2.02 | LBP-1c (leader-binding protein-1c), LSF (late SV40 factor), CP2, SEF (SAA3 enhancer factor) | 442 | (+) | 0.846 | aACTGttactttcttaata |
| CEBP.01 | CCAAT/Enhancer Binding Protein | 453 | (−) | 0.941 | tttattaaGAAAgta |
| HMX2.03 | Hmx2/Nkx5-2 homeodomain transcription factor | 466 | (−) | 0.822 | tgttctTTAAatgtgttta |
| HNF4.02 | Hepatic nuclear factor 4, DR2 sites | 519 | (−) | 0.775 | gaagttggtctAAAGaacagcttcc |
| STAT6.01 | STAT6: signal transducer and activator of transcription 6 | 529 | (−) | 0.899 | stagTTCCagagaagaagt |
| RREB1.01 | Ras-responsive element binding protein 1 | 588 | (+) | 0.840 | cCCCAaaatatccag |
| AIRE2.02 | Autoimmune regulatory element binding factor | 589 | (−) | 0.885 | actggatattTTGGg |
| BTEB3.01 | Basic transcription element (BTE) binding protein, BTEB3, FKLF-2 | 610 | (+) | 0.948 | ggaaattcagGGAGttgaa |
| RU49.04 | Zinc finger transcription factor RU49, zinc finger proliferation 1 - Zipro1 | 683 | (+) | 1.000 | aAGTAcc |
| CEBPB.01 | CCAAT/enhancer binding protein beta | 873 | (−) | 0.966 | gaaatttcGCAAtga |
| miRNA Variants | no of SNP | Position | Sequence | Accession Number | Variants IDs | |
|---|---|---|---|---|---|---|
| bta-miR-2381 | C/A C/G T/A/C T/G C/G T/G G/C | 7 | 1-19 | CAGGCUGCUCUGUGCUUGGCU | MIMAT0011929 | rs472162209 rs442506482 rs462738247 rs476414754 rs438710923 rs458831864 rs478292631 |
| bta-miR-2340 | - | - | 3-17 | GGACUUCCCUGGUGGUCUUGUG | MIMAT0011875 | N/A |
| bta-miR-12032 | T/G | 1 | 180-200 | UCUGGCCUGGAGAAGCCCUGG | MIMAT0046725 | rs438484117 |
| bta-miR-22-5p | - | - | 15-36 | AGUUCUUCAGUGGCAAGCUUUA | MIMAT0003826 | N/A |
| bta-miR-22-3p | - | - | 53-73 | AAGCUGCCAGUUGAAGAACUG | MIMAT0012536 | N/A |
| Polymorphism | Genotype | Genotypic Count (n = 103) | Frequency | Significance |
|---|---|---|---|---|
| rs721906237 | C/C | 0 | 0 | NS |
| C/A | 7 | 0.03 | * | |
| A/A | 94 | 0.97 | *** | |
| rs732566082 | G/G | 0 | 0 | NS |
| C/G | 1 | 0.01 | NS | |
| C/C | 102 | 0.99 | *** | |
| rs799300279 | G/G | 15 | 0.15 | * |
| G/A | 51 | 0.50 | *** | |
| A/A | 35 | 0.35 | ** | |
| Allele | Allelic Count | Frequency | Significance | |
| rs721906237 | C | 7 | 0.03 | *** |
| A | 195 | 0.97 | *** | |
| rs732566082 | G | 1 | 0.01 | *** |
| C | 205 | 0.99 | *** | |
| rs799300279 | G | 81 | 0.40 | *** |
| A | 121 | 0.60 | *** |
| Haplotype | Haplotype Definition | Haplotype Frequency | p Value | ||
|---|---|---|---|---|---|
| rs721906237 (C/A) | rs723566082 (G/C) | rs799300279 (A/G) | |||
| H1 | A | C | A | 0.5594 | 0.002 |
| H2 | A | C | G | 0.4012 | 0.046 |
| H3 | C | C | A | 0.0346 | 0.990 |
| H4 | A | G | A | 0.0049 | 0.068 |
| H5 | C | C | G | 0 | NS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morenikeji, O.B.; Capria, A.L.; Ojurongbe, O.; Thomas, B.N. SNP Diversity in CD14 Gene Promoter Suggests Adaptation Footprints in Trypanosome Tolerant N’Dama (Bos taurus) but not in Susceptible White Fulani (Bos indicus) Cattle. Genes 2020, 11, 112. https://doi.org/10.3390/genes11010112
Morenikeji OB, Capria AL, Ojurongbe O, Thomas BN. SNP Diversity in CD14 Gene Promoter Suggests Adaptation Footprints in Trypanosome Tolerant N’Dama (Bos taurus) but not in Susceptible White Fulani (Bos indicus) Cattle. Genes. 2020; 11(1):112. https://doi.org/10.3390/genes11010112
Chicago/Turabian StyleMorenikeji, Olanrewaju B., Anna L. Capria, Olusola Ojurongbe, and Bolaji N. Thomas. 2020. "SNP Diversity in CD14 Gene Promoter Suggests Adaptation Footprints in Trypanosome Tolerant N’Dama (Bos taurus) but not in Susceptible White Fulani (Bos indicus) Cattle" Genes 11, no. 1: 112. https://doi.org/10.3390/genes11010112
APA StyleMorenikeji, O. B., Capria, A. L., Ojurongbe, O., & Thomas, B. N. (2020). SNP Diversity in CD14 Gene Promoter Suggests Adaptation Footprints in Trypanosome Tolerant N’Dama (Bos taurus) but not in Susceptible White Fulani (Bos indicus) Cattle. Genes, 11(1), 112. https://doi.org/10.3390/genes11010112