Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland

 ,
,  , ,
, ,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Identification and Recruitment
2.2. DNA Acquisition and Next Generation Sequencing
2.3. Variant Confirmation Sequencing
2.4. Choroideremia Deletion Confirmation
2.5. Sequencing of RPGR ORF15
2.6. Single-Molecule Molecular Inversion Probe (smMIP)-Based Sequencing of ABCA4
2.7. Data Analysis and Variant Interpretation of Target Capture NGS Data
2.8. Ethical Approval
3. Results
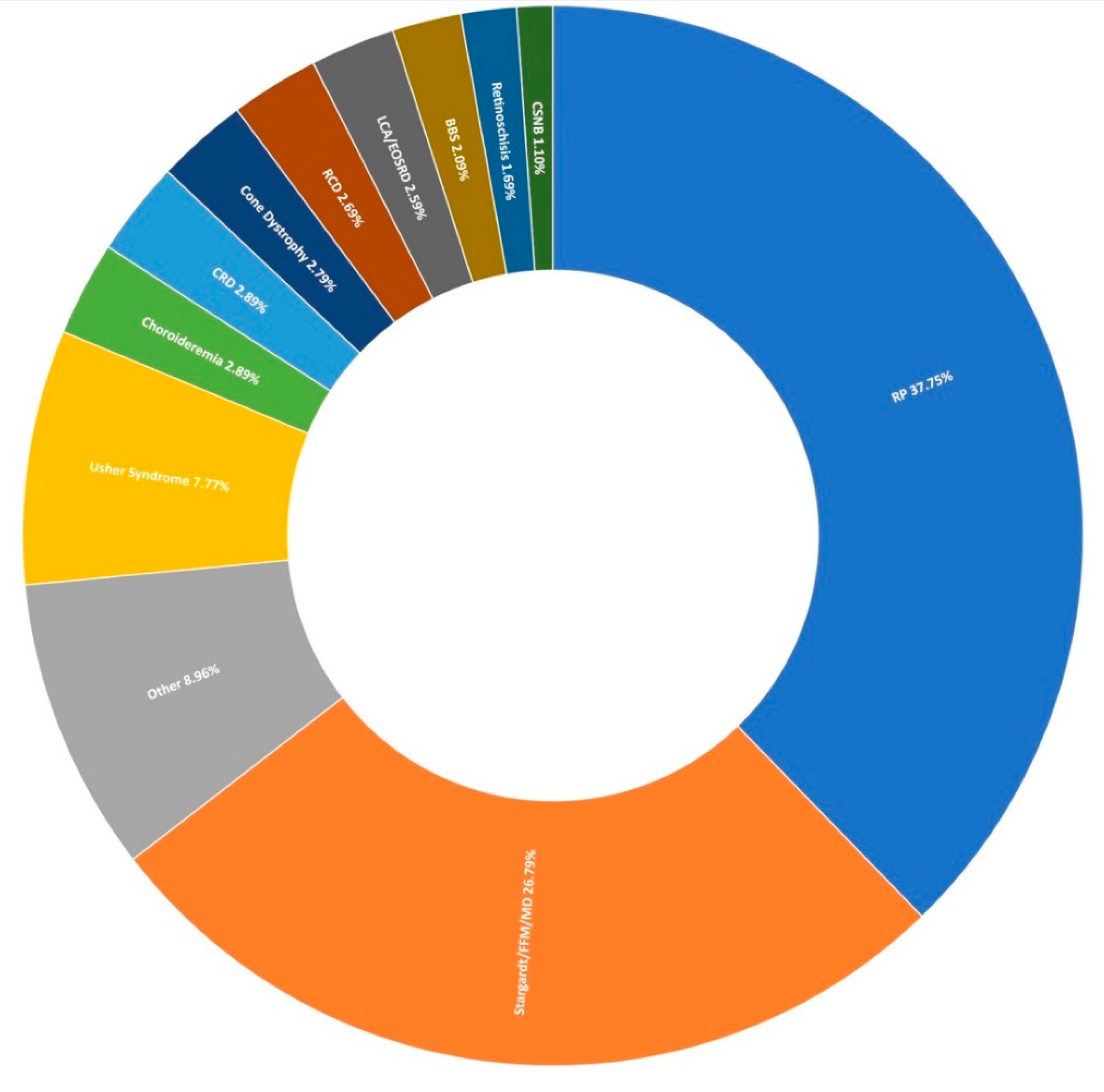
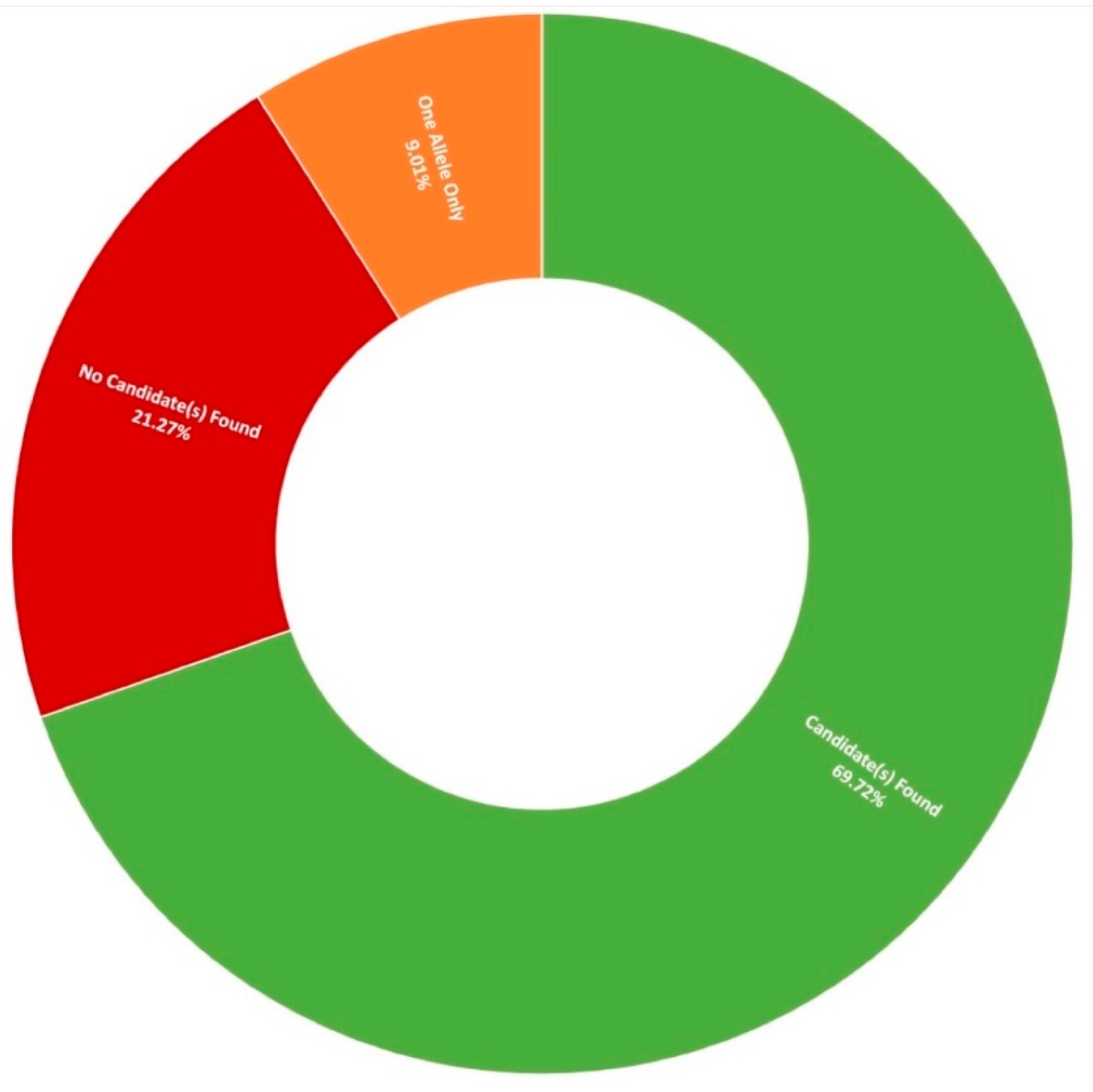
3.1. Clinical Presentation and Positive Candidate Detection Rates
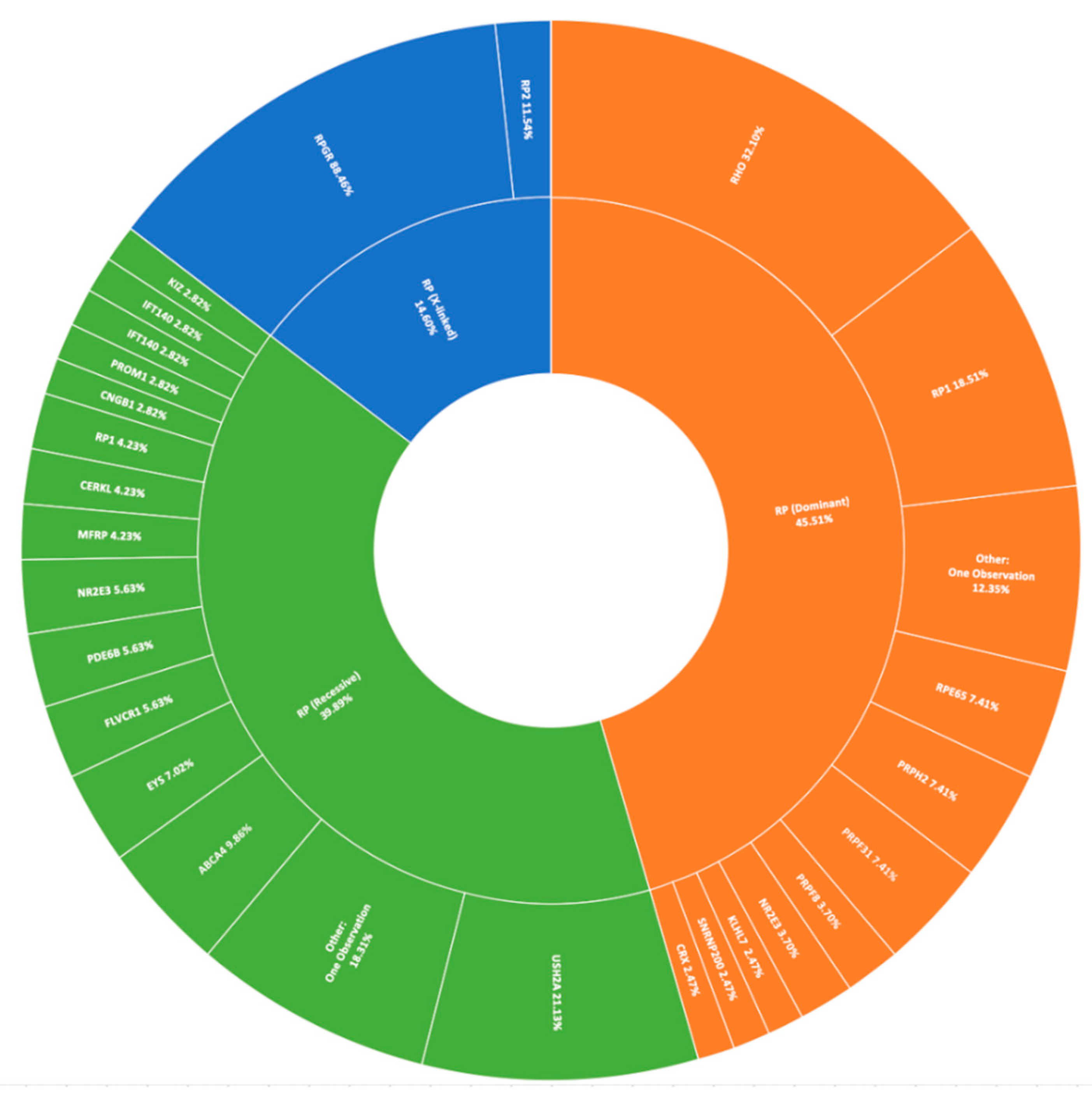
3.2. Retinitis Pigmentosa
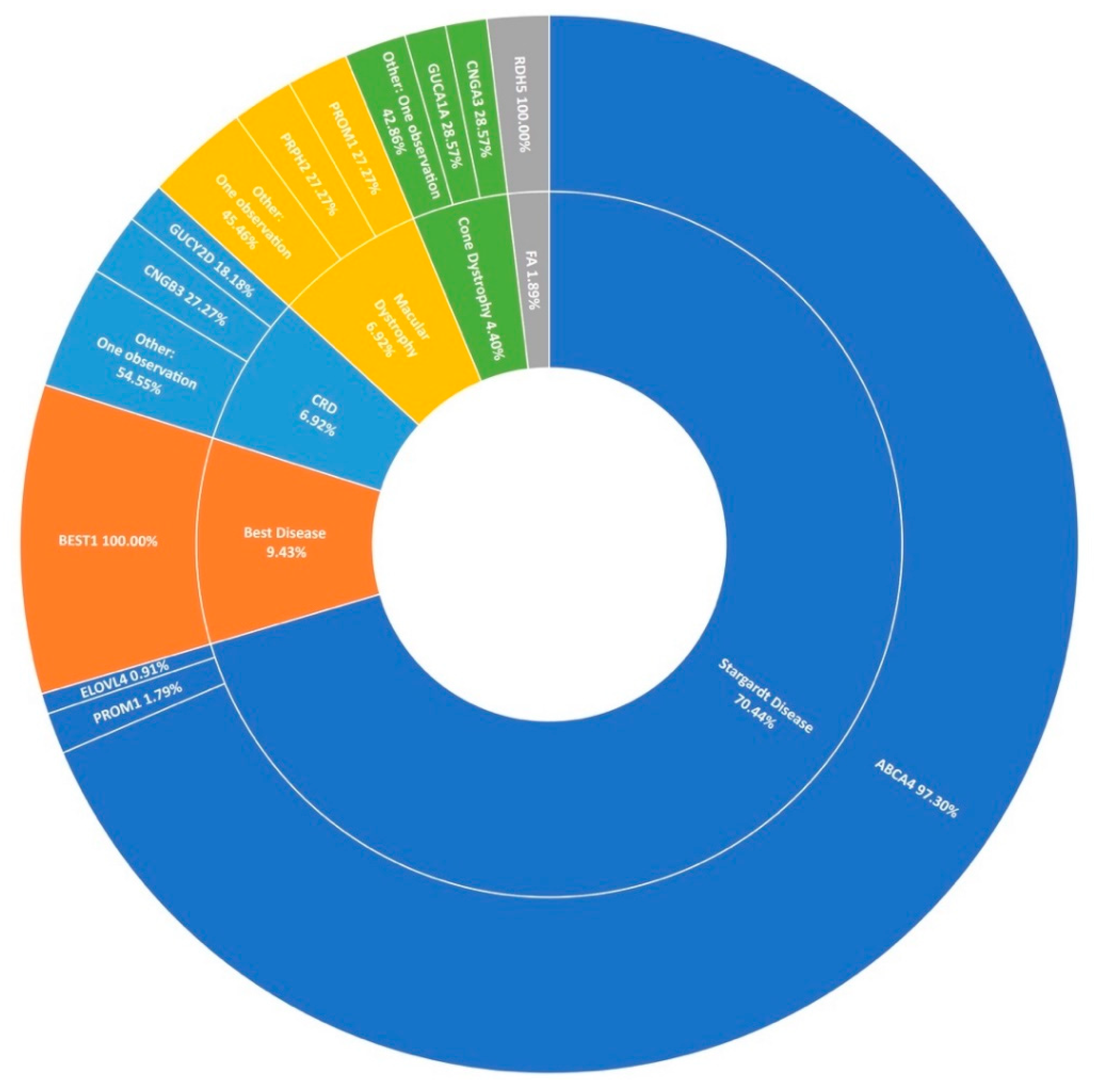
3.3. Stargardt Disease and Other Macular Dystrophies
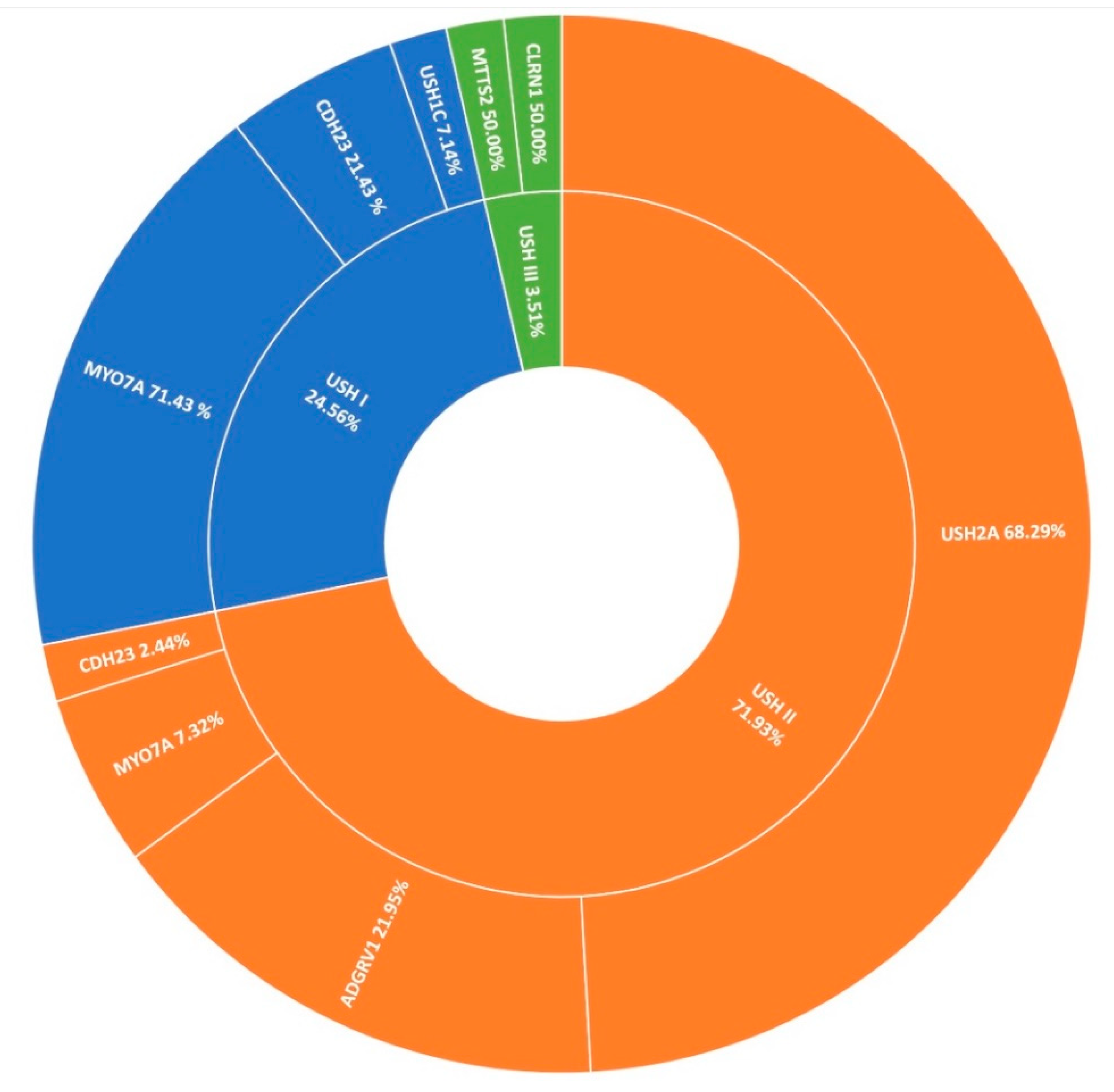
3.4. Usher Syndrome
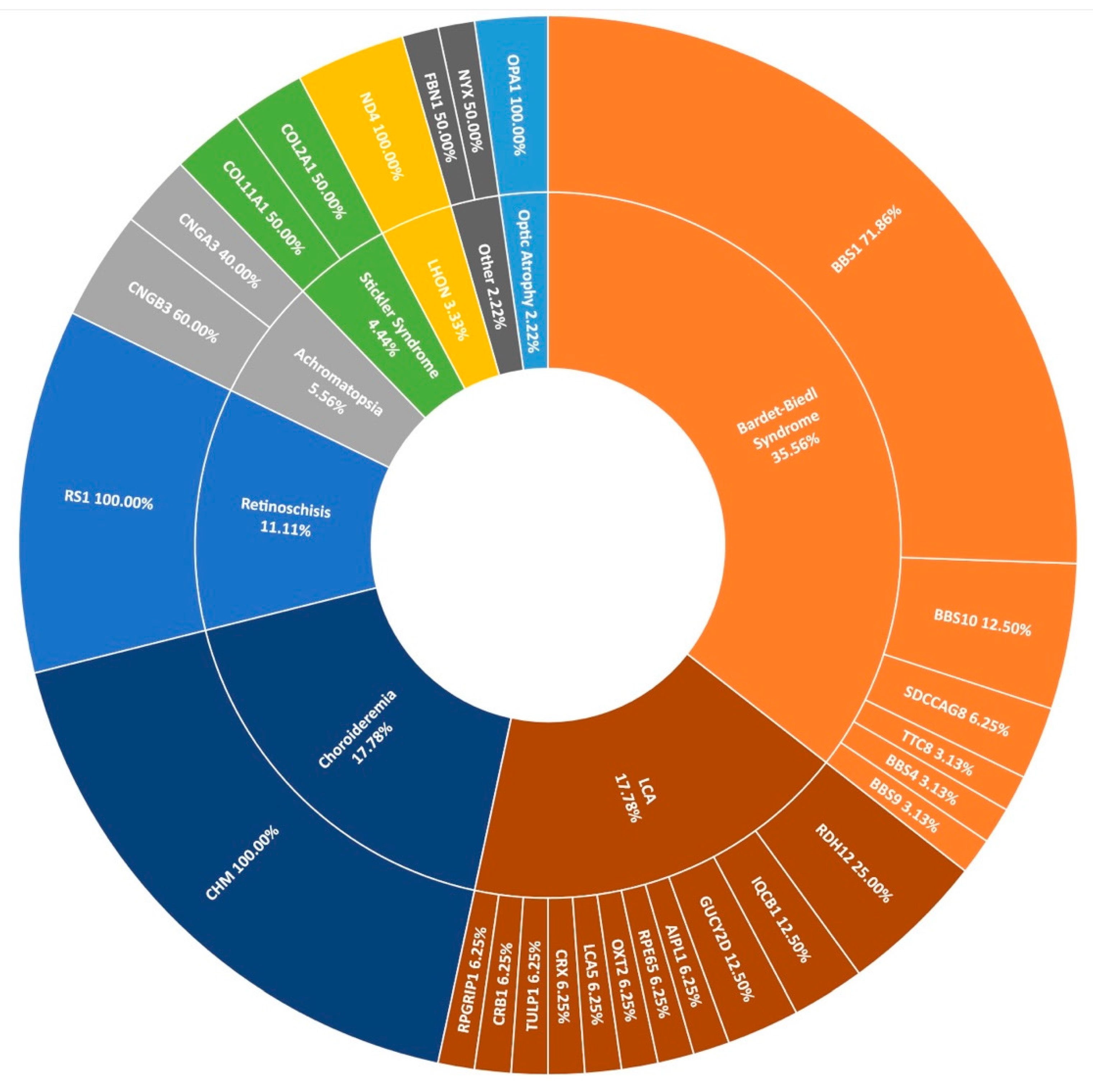
3.5. Other IRDs Encompassed by Target 5000
3.6. Novel Variants
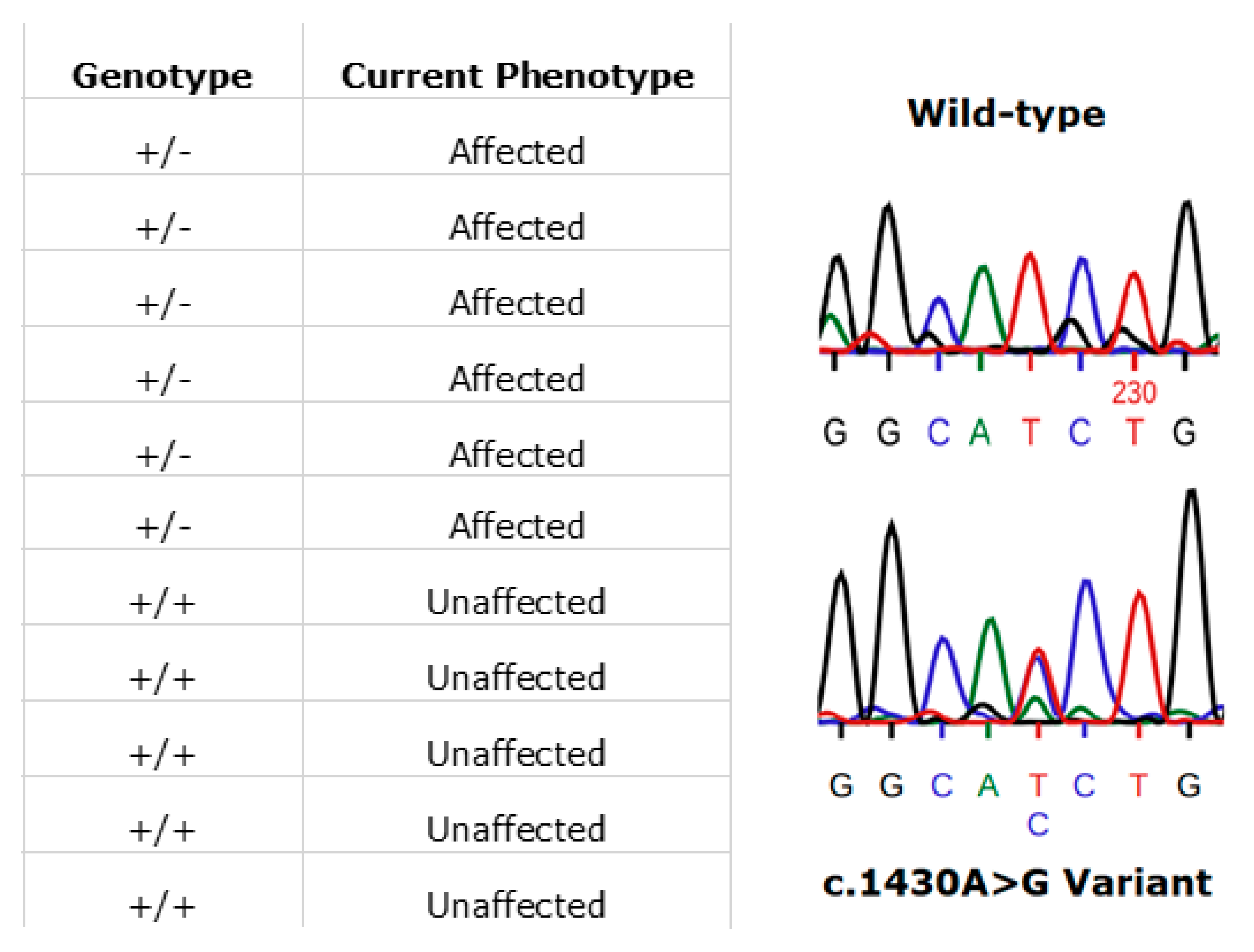
3.7. RPE65
3.8. FLVCR1
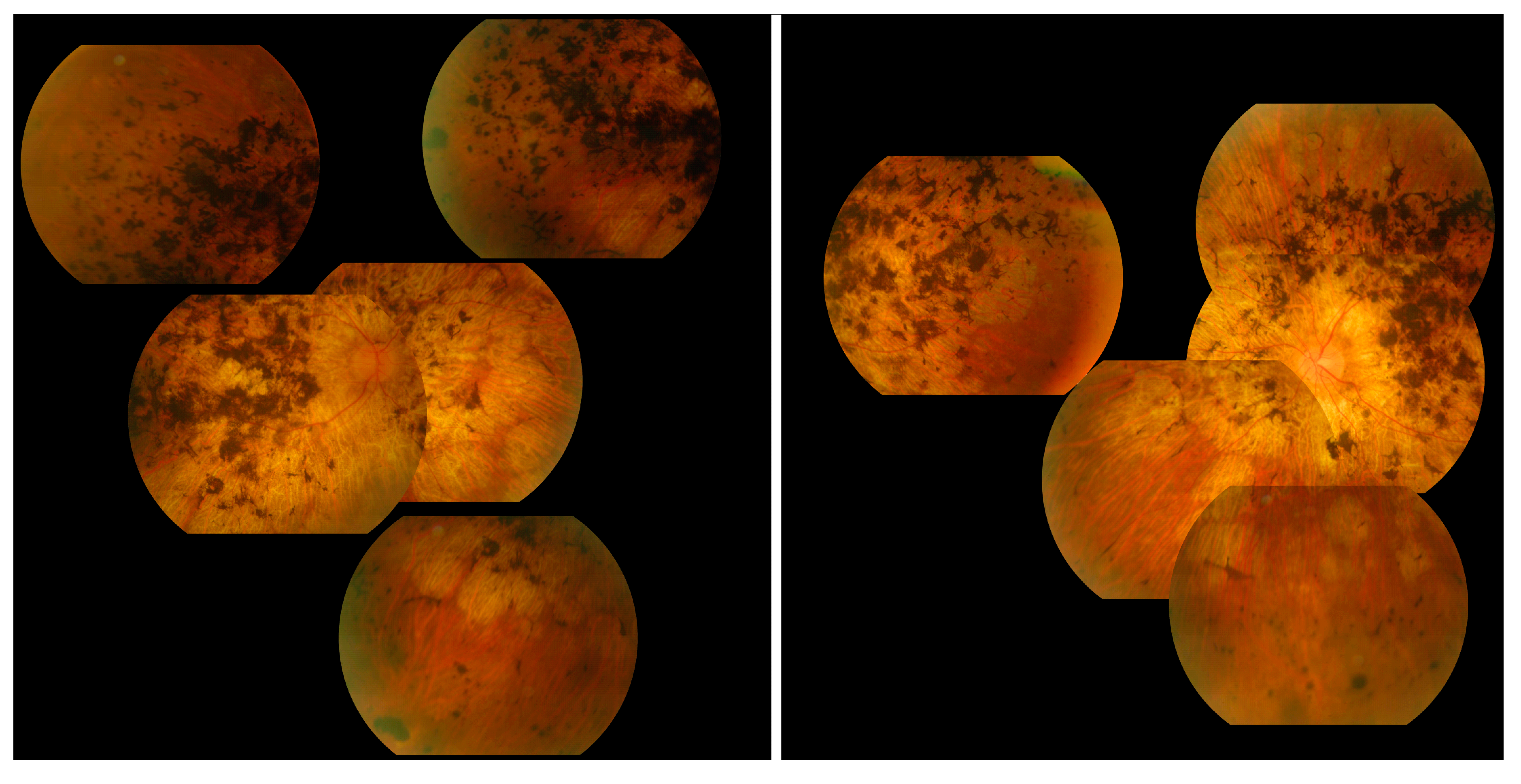
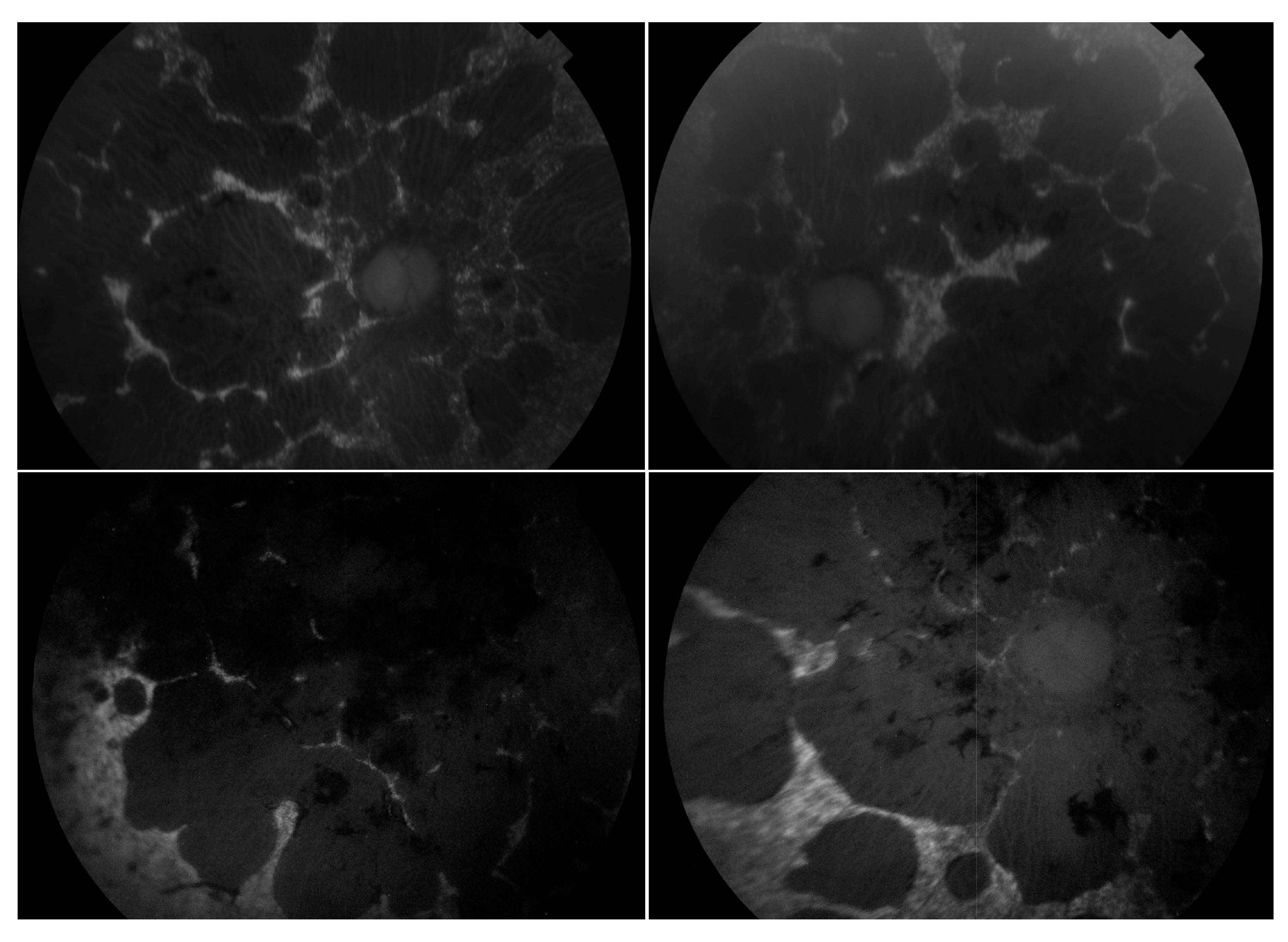
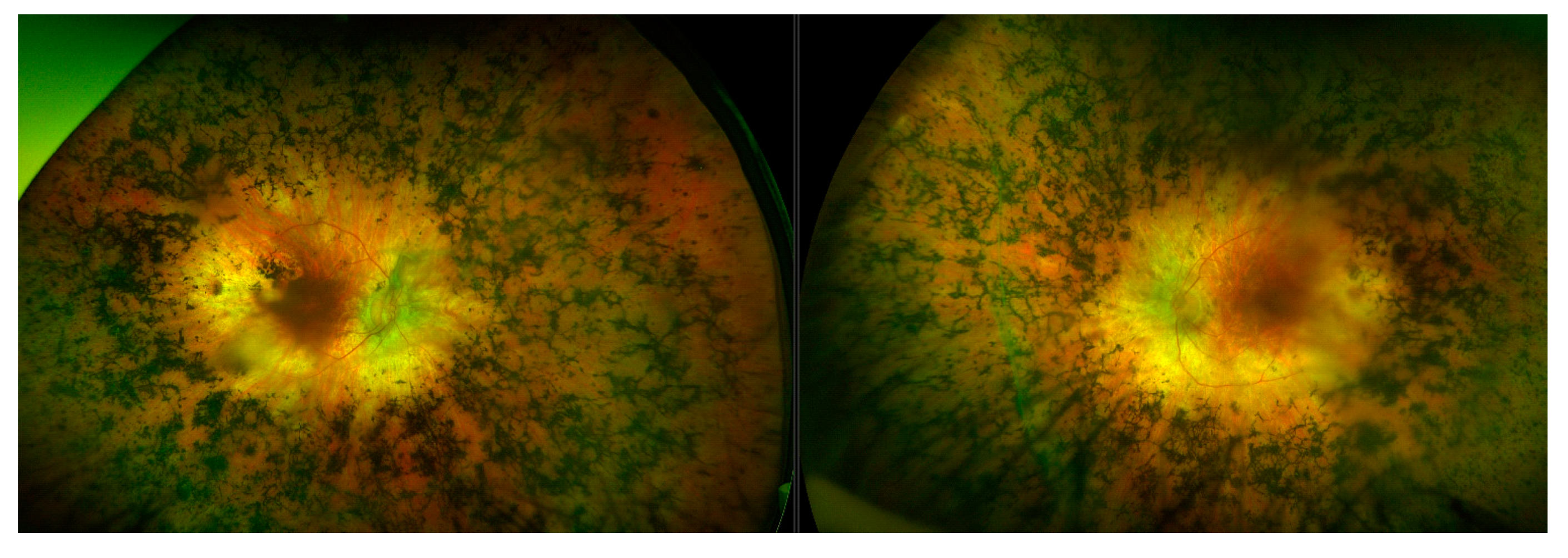
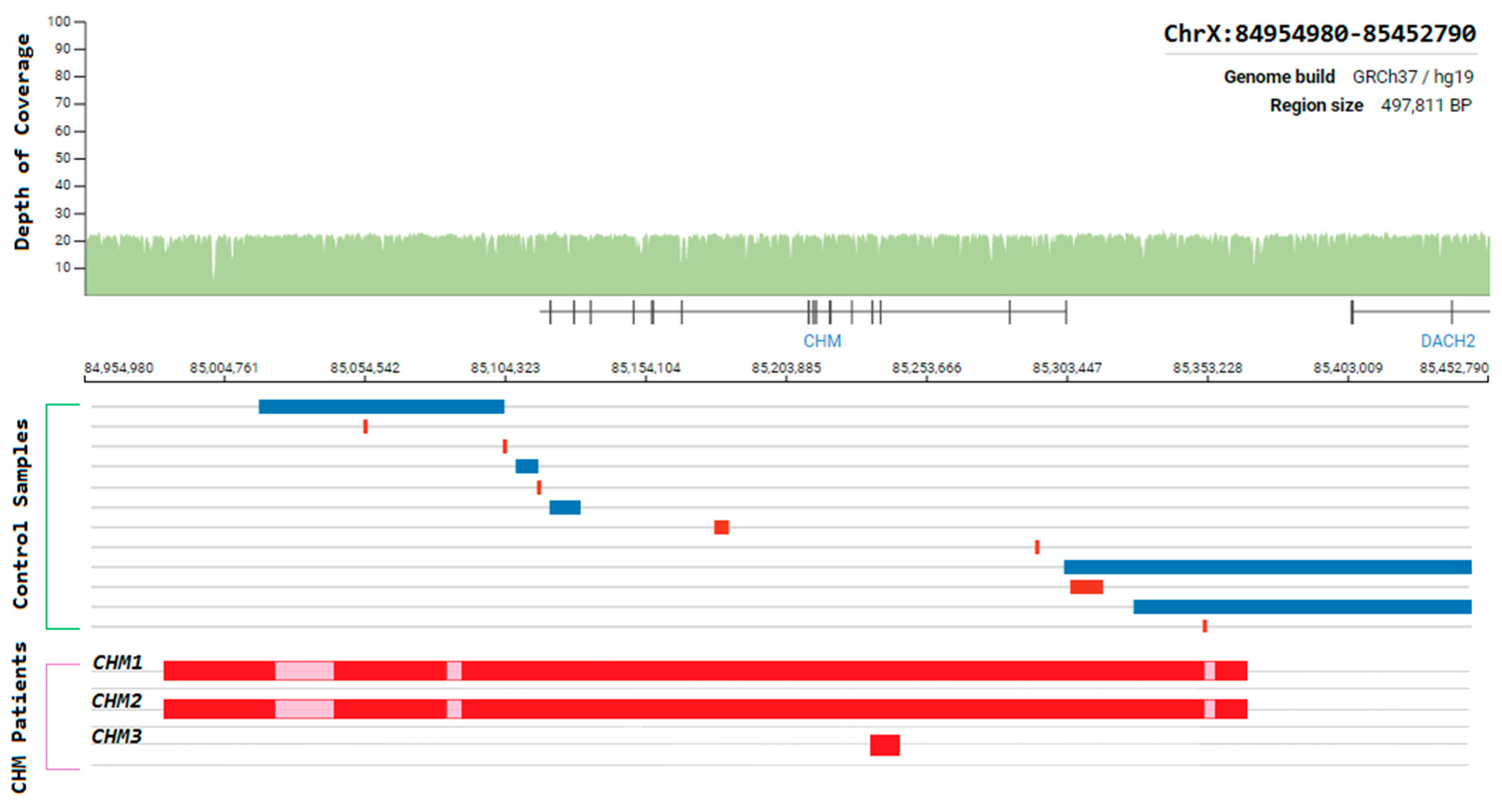
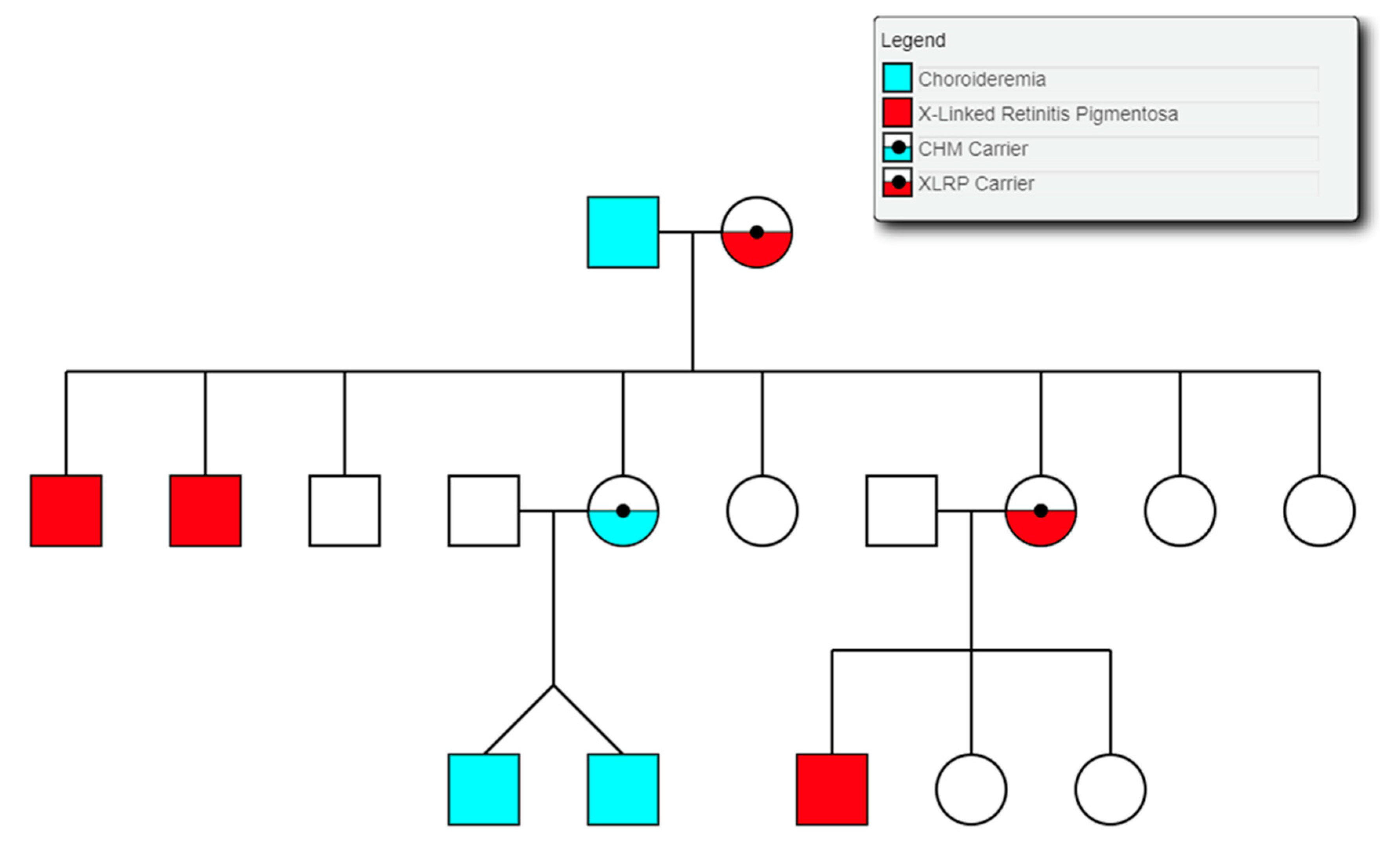
3.9. Choroideremia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mei, S.; Huang, X.; Cheng, L.; Peng, S.; Zhu, T.; Chen, L.; Wang, Y.; Zhao, J. A missense mutation in OPA1 causes dominant optic atrophy in a Chinese family. J. Ophthalmol. 2019, 2019, 1424928. [Google Scholar] [CrossRef]
- Gao, F.-J.; Zhang, S.-H.; Chen, J.-Y.; Xu, G.-Z.; Wu, J.-H. Digenic heterozygous mutations in EYS/LRP5 in a Chinese family with retinitis pigmentosa. Int. J. Ophthalmol. 2017, 10, 325–328. [Google Scholar]
- Bhattacharya, S.S.; Wright, A.F.; Clayton, J.F.; Price, W.H.; Phillips, C.I.; McKeown, C.M.; Jay, M.; Bird, A.C.; Pearson, P.L.; Southern, E.M. Close genetic linkage between X-linked retinitis pigmentosa and a restriction fragment length polymorphism identified by recombinant DNA probe L1.28. Nature 1984, 309, 253–255. [Google Scholar] [CrossRef]
- Farrar, G.J.; Kenna, P.; Jordan, S.A.; Kumar-Singh, R.; Humphries, M.M.; Sharp, E.M.; Sheils, D.M.; Humphries, P. A three-base-pair deletion in the peripherin–RDS gene in one form of retinitis pigmentosa. Nature 1991, 354, 478–480. [Google Scholar] [CrossRef]
- Kumar-Singh, R.; Bradley, D.G.; Farrar, G.J.; Lawler, M.; Jordan, S.A.; Humphries, P. Autosomal dominant retinitis pigmentosa: A new multi-allelic marker (D3S621) genetically linked to the disease locus (RP4). Hum. Genet. 1991, 86, 502–504. [Google Scholar] [CrossRef] [PubMed]
- RetNet: Summaries. Available online: https://sph.uth.edu/retnet/sum-dis.htm (accessed on 29 October 2019).
- Werdich, X.Q.; Place, E.M.; Pierce, E.A. Systemic diseases associated with retinal dystrophies. Semin. Ophthalmol. 2014, 29, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genomics 2011, 12, 238–249. [Google Scholar] [PubMed]
- Farrar, G.J.; Carrigan, M.; Dockery, A.; Millington-Ward, S.; Palfi, A.; Chadderton, N.; Humphries, M.; Kiang, A.S.; Kenna, P.F.; Humphries, P. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum. Mol. Genet. 2017, 26, R2–R11. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef]
- Maugeri, A.; Klevering, B.J.; Rohrschneider, K.; Blankenagel, A.; Brunner, H.G.; Deutman, A.F.; Hoyng, C.B.; Cremers, F.P.M. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am. J. Hum. Genet. 2000, 67, 960–966. [Google Scholar] [CrossRef]
- Maugeri, A.; van Driel, M.A.; van de Pol, D.J.; Klevering, B.J.; van Haren, F.J.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; Pinckers, A.J.; et al. The 2588G→C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease. Am. J. Hum. Genet. 1999, 64, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Pozo-Valero, M.; del Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTabishi, A.; Baere, E.D.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. bioRxiv 2019, 817767. [Google Scholar] [CrossRef]
- Kim, D.Y.; Mukai, S. X-linked Juvenile Retinoschisis (XLRS): A review of genotype-phenotype relationships. Semin. Ophthalmol. 2013, 28, 392–396. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Stephenson, K.; Keegan, D.; Wynne, N.; Silvestri, G.; Humphries, P.; Kenna, P.F.; Carrigan, M.; Farrar, G.J. Target 5000: Target capture sequencing for inherited retinal degenerations. Genes 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.M.; Russell, S.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; Marshall, K.A.; et al. Efficacy, Safety, and Durability of Voretigene Neparvovec-rzyl in RPE65 Mutation-Associated Inherited Retinal Dystrophy: Results of Phase 1 and 3 Trials. Ophthalmology 2019, 126, 1273–1285. [Google Scholar] [CrossRef]
- Consugar, M.B.; Navarro-Gomez, D.; Place, E.M.; Bujakowska, K.M.; Sousa, M.E.; Fonseca-Kelly, Z.D.; Taub, D.G.; Janessian, M.; Wang, D.Y.; Au, E.D.; et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med. 2015, 17, 253–261. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Li, J.; Tang, J.; Feng, Y.; Xu, M.; Chen, R.; Zou, X.; Sui, R.; Chang, E.Y.; Lewis, R.A.; Zhang, V.W.; et al. Improved diagnosis of inherited retinal dystrophies by high-fidelity PCR of ORF15 followed by next-generation sequencing. J. Mol. Diagn. 2016, 18, 817–824. [Google Scholar] [CrossRef]
- Boyle, E.A.; O’Roak, B.J.; Martin, B.K.; Kumar, A.; Shendure, J. MIPgen: Optimized modeling and design of molecular inversion probes for targeted resequencing. Bioinformatics 2014, 30, 2670–2672. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Jarvik, G.P.; Browning, B.L. Consideration of cosegregation in the pathogenicity classification of genomic variants. Am. J. Hum. Genet. 2016, 98, 1077–1081. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Odou, M.-F.; North, M.-O.; Saveanu, A.; Coppin, L.; Pasmant, E.; Mohamed, A.; Goudet, P.; Borson-Chazot, F.; Calender, A.; et al. Proposition of adjustments to the ACMG-AMP framework for the interpretation of MEN1 missense variants. Hum. Mutat. 2019, 40, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Jian, X.; Liu, X. In silico prediction of deleteriousness for nonsynonymous and splice-altering single nucleotide variants in the human genome. In Vitro Mutagenesis; Reeves, A., Ed.; Springer: New York, NY, USA, 2017; Volume 1498, pp. 191–197. ISBN 978-1-4939-6470-3. [Google Scholar]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef]
- Carrigan, M.; Duignan, E.; Malone, C.P.G.; Stephenson, K.; Saad, T.; McDermott, C.; Green, A.; Keegan, D.; Humphries, P.; Kenna, P.F.; et al. Panel-based population next-generation sequencing for inherited retinal degenerations. Sci. Rep. 2016, 6, 33248. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of disease-causing mutations in families with autosomal dominant retinitis pigmentosa: A screen of known genes in 200 families. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef]
- Lenassi, E.; Vincent, A.; Li, Z.; Saihan, Z.; Coffey, A.J.; Steele-Stallard, H.B.; Moore, A.T.; Steel, K.P.; Luxon, L.M.; Héon, E.; et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur. J. Hum. Genet. 2015, 23, 1318–1327. [Google Scholar] [CrossRef]
- Klevering, B.J.; Yzer, S.; Rohrschneider, K.; Zonneveld, M.; Allikmets, R.; van den Born, L.I.; Maugeri, A.; Hoyng, C.B.; Cremers, F.P.M. Microarray-based mutation analysis of the ABCA4 (ABCR) gene in autosomal recessive cone-rod dystrophy and retinitis pigmentosa. Eur. J. Hum. Genet. 2004, 12, 1024–1032. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; van den Born, L.I.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; Vorst, M.; van de Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- GnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 14 November 2019).
- Bowne, S.J.; Humphries, M.M.; Sullivan, L.S.; Kenna, P.F.; Tam, L.C.S.; Kiang, A.S.; Campbell, M.; Weinstock, G.M.; Koboldt, D.C.; Ding, L.; et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur. J. Hum. Genet. 2011, 19, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Hull, S.; Mukherjee, R.; Holder, G.E.; Moore, A.T.; Webster, A.R. The clinical features of retinal disease due to a dominant mutation in RPE65. Mol. Vis. 2016, 22, 626–635. [Google Scholar]
- Li, S.; Xiao, X.; Yi, Z.; Sun, W.; Wang, P.; Zhang, Q. RPE65 mutation frequency and phenotypic variation according to exome sequencing in a tertiary centre for genetic eye diseases in China. Acta Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Jauregui, R.; Park, K.S.; Tsang, S.H. Two-year progression analysis of RPE65 autosomal dominant retinitis pigmentosa. Ophthalmic Genet. 2018, 39, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the heme exporter FLVCR1 cause sensory neurodegeneration with loss of pain perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef]
- Glöckle, N.; Kohl, S.; Mohr, J.; Scheurenbrand, T.; Sprecher, A.; Weisschuh, N.; Bernd, A.; Rudolph, G.; Schubach, M.; Poloschek, C.; et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur. J. Hum. Genet. 2014, 22, 99–104. [Google Scholar] [CrossRef]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed]
- Dockery, A.; Carrigan, M.; Wynne, N.; Stephenson, K.; Keegan, D.; Kenna, P.F.; Farrar, G.J. A novel FLVCR1 variant implicated in retinitis pigmentosa. In Retinal Degenerative Diseases; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2019; Volume 1185. [Google Scholar]
- Patel, A.; Hayward, J.D.; Tailor, V.; Nyanhete, R.; Ahlfors, H.; Gabriel, C.; Jannini, T.B.; Abbou-Rayyah, Y.; Henderson, R.; Nischal, K.K.; et al. The Oculome panel test: Next-generation sequencing to diagnose a diverse range of genetic developmental eye disorders. Ophthalmology 2019, 126, 888–907. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef] [PubMed]
- Riera, M.; Navarro, R.; Ruiz-Nogales, S.; Méndez, P.; Burés-Jelstrup, A.; Corcóstegui, B.; Pomares, E. Whole exome sequencing using Ion Proton system enables reliable genetic diagnosis of inherited retinal dystrophies. Sci. Rep. 2017, 7, 42078. [Google Scholar] [CrossRef]
- Sharon, D.; Ben-Yosef, T.; Goldenberg-Cohen, N.; Pras, E.; Gradstein, L.; Soudry, S.; Mezer, E.; Zur, D.; Abbasi, A.H.; Zeitz, C.; et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum. Mutat. 2020, 41, 140–149. [Google Scholar] [CrossRef]
- Haer-Wigman, L.; van Zelst-Stams, W.A.; Pfundt, R.; van den Born, L.I.; Klaver, C.C.; Verheij, J.B.; Hoyng, C.B.; Breuning, M.H.; Boon, C.J.; Kievit, A.J.; et al. Diagnostic exome sequencing in 266 Dutch patients with visual impairment. Eur. J. Hum. Genet. 2017, 25, 591–599. [Google Scholar] [CrossRef]
- Ellingford, J.M.; Barton, S.; Bhaskar, S.; Williams, S.G.; Sergouniotis, P.I.; O’Sullivan, J.; Lamb, J.A.; Perveen, R.; Hall, G.; Newman, W.G.; et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology 2016, 123, 1143–1150. [Google Scholar] [CrossRef]
- Allikmets, R.; Shroyer, N.F.; Singh, N.; Seddon, J.M.; Lewis, R.A.; Bernstein, P.S.; Peiffer, A.; Zabriskie, N.A.; Li, Y.; Hutchinson, A.; et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997, 277, 1805–1807. [Google Scholar] [CrossRef]
- Strauss, R.W.; Ho, A.; Muñoz, B.; Cideciyan, A.V.; Sahel, J.-A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies: Design and baseline characteristics: ProgStar Report No. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef]
- Fishman, G.A.; Farbman, J.S.; Alexander, K.R. Delayed rod dark adaptation in patients with Stargardt’s disease. Ophthalmology 1991, 98, 957–962. [Google Scholar] [CrossRef]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; den Dunnen, J.T.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P.M. In silico functional meta-analysis of 5,962 ABCA4 variants in 3,928 retinal dystrophy cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.J.; et al. Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Edwards, P.C.; Burghammer, M.; Villa, C.; Schertler, G.F.X. Structure of bovine rhodopsin in a trigonal crystal form. J. Mol. Biol. 2004, 343, 1409–1438. [Google Scholar] [CrossRef] [PubMed]
- Keel, S.B.; Doty, R.T.; Yang, Z.; Quigley, J.G.; Chen, J.; Knoblaugh, S.; Kingsley, P.D.; De Domenico, I.; Vaughn, M.B.; Kaplan, J.; et al. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science 2008, 319, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Tailor, C.S.; Willett, B.J.; Kabat, D. A putative cell surface receptor for anemia-inducing feline leukemia virus subgroup C is a member of a transporter superfamily. J. Virol. 1999, 73, 6500–6505. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, S.R.; Fitzpatrick, Z.; Harris, A.F.; Maitland, S.A.; Ferreira, J.S.; Zhang, Y.; Ma, S.; Sharma, R.B.; Gray-Edwards, H.L.; Johnson, J.A.; et al. In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol. Ther. 2016, 24, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, A.-K.; Grosse, S.; Börner, K.; Krämer, C.; Wiedtke, E.; Gunkel, M.; Grimm, D. Impact of the assembly-activating protein (AAP) on molecular evolution of synthetic Adeno-associated virus (AAV) capsids. Hum. Gene Ther. 2018, 30, 21–35. [Google Scholar] [CrossRef]
- Tordo, J.; O’Leary, C.; Antunes, A.S.L.M.; Palomar, N.; Aldrin-Kirk, P.; Basche, M.; Bennett, A.; D’Souza, Z.; Gleitz, H.; Godwin, A.; et al. A novel adeno-associated virus capsid with enhanced neurotropism corrects a lysosomal transmembrane enzyme deficiency. Brain J. Neurol. 2018, 141, 2014–2031. [Google Scholar] [CrossRef]
- Smith, J.K.; Agbandje-McKenna, M. Creating an arsenal of Adeno-associated virus (AAV) gene delivery stealth vehicles. PLoS Pathog. 2018, 14, e1006929. [Google Scholar] [CrossRef]
- Paulk, N.K.; Pekrun, K.; Zhu, E.; Nygaard, S.; Li, B.; Xu, J.; Chu, K.; Leborgne, C.; Dane, A.P.; Haft, A.; et al. Bioengineered AAV capsids with combined high human liver transduction in vivo and unique humoral seroreactivity. Mol. Ther. 2018, 26, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Marlhens, F.; Bareil, C.; Griffoin, J.-M.; Zrenner, E.; Amalric, P.; Eliaou, C.; Liu, S.-Y.; Harris, E.; Redmond, T.M.; Arnaud, B.; et al. Mutations in RPE65 cause Leber’s congenital amaurosis. Nat. Genet. 1997, 17, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Marlhens, F.; Griffoin, J.-M.; Bareil, C.; Arnaud, B.; Claustres, M.; Hamel, C.P. Autosomal recessive retinal dystrophy associated with two novel mutations in the RPE65 gene. Eur. J. Hum. Genet. 1998, 6, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P.; Griffoin, J.-M.; Lasquellec, L.; Bazalgette, C.; Arnaud, B. Retinal dystrophies caused by mutations in RPE65: Assessment of visual functions. Br. J. Ophthalmol. 2001, 85, 424–427. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, E.H.; Suh, S.; Sander, C.L.; Hernandez, C.J.O.; Bulman, E.R.; Khadka, N.; Dong, Z.; Shi, W.; Palczewski, K.; Kiser, P.D. Insights into the pathogenesis of dominant retinitis pigmentosa associated with a D477G mutation in RPE65. Hum. Mol. Genet. 2018, 27, 2225–2243. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Izumi, T.; Hu, J.; Jin, H.H.; Siddiqui, A.-A.A.; Jacobson, S.G.; Bok, D.; Jin, M. Rescue of enzymatic function for disease-associated RPE65 proteins containing various missense mutations in non-active sites. J. Biol. Chem. 2014, 289, 18943–18956. [Google Scholar] [CrossRef]
- Li, Y.; Furhang, R.; Ray, A.; Duncan, T.; Soucy, J.; Mahdi, R.; Chaitankar, V.; Gieser, L.; Poliakov, E.; Qian, H.; et al. Aberrant RNA splicing is the major pathogenic effect in a knock-in mouse model of the dominantly inherited c.1430A>G human RPE65 mutation. Hum. Mutat. 2019, 40, 426–443. [Google Scholar] [CrossRef]
- Simunovic, M.P.; Jolly, J.K.; Xue, K.; Edwards, T.L.; Groppe, M.; Downes, S.M.; MacLaren, R.E. The spectrum of CHM gene mutations in choroideremia and their relationship to clinical phenotype. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6033–6039. [Google Scholar] [CrossRef]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef]
- Radziwon, A.; Arno, G.K.; Wheaton, D.; McDonagh, E.M.; Baple, E.L.; Webb-Jones, K.G.; Birch, D.; Webster, A.R.; MacDonald, I.M. Single-base substitutions in the CHM promoter as a cause of choroideremia. Hum. Mutat. 2017, 38, 704–715. [Google Scholar] [CrossRef]
- Van Schil, K.; Naessens, S.; Van de Sompele, S.; Carron, M.; Aslanidis, A.; Van Cauwenbergh, C.; Kathrin Mayer, A.; Van Heetvelde, M.; Bauwens, M.; Verdin, H.; et al. Mapping the genomic landscape of inherited retinal disease genes prioritizes genes prone to coding and noncoding copy-number variations. Genet. Med. 2018, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Uppal, S.; Liu, T.; Poliakov, E.; Gentleman, S.; Redmond, T.M. The dual roles of RPE65 S-palmitoylation in membrane association and visual cycle function. Sci. Rep. 2019, 9, 5218. [Google Scholar] [CrossRef] [PubMed]
- Ellingford, J.M.; Thomas, H.B.; Rowlands, C.; Arno, G.; Beaman, G.; Gomes-Silva, B.; Campbell, C.; Gossan, N.; Hardcastle, C.; Webb, K.; et al. Functional and in-silico interrogation of rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. bioRxiv 2019, 781088. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Fadaie, Z.; Klevering, B.J.; van Genderen, M.M.; Feenstra, I.; Cremers, F.P.M.; Hoyng, C.B.; Roosing, S. The identification of a RNA splice variant in TULP1 in two siblings with early-onset photoreceptor dystrophy. Mol. Genet. Genomic Med. 2019, 7, e660. [Google Scholar] [CrossRef]
- Dickinson, D.J.; Goldstein, B. CRISPR-based methods for Caenorhabditis elegans genome engineering. Genetics 2016, 202, 885–901. [Google Scholar] [CrossRef]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. [Google Scholar] [CrossRef]
- Dam, T.J.P.; van Kennedy, J.; Lee, R.; van der Vrieze, E.; de Wunderlich, K.A.; Rix, S.; Dougherty, G.W.; Lambacher, N.J.; Li, C.; Jensen, V.L.; et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PLoS ONE 2019, 14, e0216705. [Google Scholar]
- Messchaert, M.; Dona, M.; Broekman, S.; Peters, T.A.; Corral-Serrano, J.C.; Slijkerman, R.W.N.; van Wijk, E.; Collin, R.W.J. Eyes shut homolog is important for the maintenance of photoreceptor morphology and visual function in zebrafish. PLoS ONE 2018, 13, e0200789. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyses | Method | Manufacturer |
|---|---|---|
| Best-corrected visual acuity | Revised 2000 early treatment diabetic retinopathy study (ETDRS) charts | Precision Vision, La Salle, IL, USA |
| Colour vision | Lanthony desaturated D-15 panel under standardised lighting conditions | Gulden Ophthalmics, Elkins Park, PA, USA |
| Peripheral visual fields | Goldmann perimeter 940 (iv4e, i4e and 04e targets) | Haag-Streit AG, Köniz, Switzerland |
| Full field electroretinograms | Roland Consult RETI-port retiscan. | Brandenburg an der Havel, Germany |
| Fundus colour and autofluorescence photography | Topcon CRC50DX/Optos Daytona | Topcon Great Britain Ltd., Berkshire, England/Optos plc, Dunfermline, Scotland |
| Spectral domain optical coherence tomography | Cirrus HD-OCT | Carl Zeiss Meditec, Berlin, Germany |
| Gene | Condition | Transcript ID | Chr | Genomic Location | Nucleotide Change | Protein Change | ACMG Criteria | ACMG Classification | MetaLR Score | M-CAP Score | REVEL Score | Observed With |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ABCA4 | Stargardt Disease | NM_000350.2 | chr1 | g.94062649 | c.1865G>A | p.Ser622Asn | PM2, PP3, PP4(S) | Likely Pathogenic | 0.668 | 0.065 | 0.279 | c.161G>A |
| ABCA4 | Macular Dystophy (Recessive) | NM_000350.2 | chr1 | g.94111517 | c.223T>C | p.Cys75Arg | PM2, PM5, PP3 | VUS | 0.995 | 0.711 | 0.967 | c.4253+43G>A |
| ABCA4 | Stargardt Disease | NM_000350.2 | chr1 | g.94029516 | c.4468T>C | p.Cys1490Arg | PM2, PP3, PP4(S) | Likely Pathogenic | 0.952 | 0.723 | 0.932 | c.3305A>T |
| ABCA4 | Macular Dystophy (Recessive) | NM_000350.2 | chr1 | g.94014674 | c.5329A>G | p.Met1777Val | PM2, PP3 | VUS | 0.663 | 0.066 | 0.758 | c.1037A>C |
| ABCA4 | Stargardt Disease | NM_000350.2 | chr1 | g.93996182 | c.6743T>C | p.Phe2248Ser | PM2, PP3, PP4(S) | Likely Pathogenic | 0.861 | 0.528 | 0.935 | c.302+68C>T |
| AHI1 | Retinitis Pigmentosa (Recessive) | NM_001134830.1 | chr6 | g.135428670 | c.2582G>A | p.Gly861Glu | PM2, PM3, PP1, PP3 | Likely Pathogenic | 0.583 | 0.159 | 0.816 | c.1267C>T |
| AIPL1 | Leber Congenital Amaurosis | NM_014336.4 | chr17 | g.6425759 | c.856G>C | p.Asp286His | PM2, PM3, PP1, PP3 | Likely Pathogenic | 0.877 | 0.290 | 0.791 | c.834G>A |
| BBS10 | Retinitis Pigmentosa (Recessive) | NM_024685.3 | chr12 | g.76348204 | c.155G>A | p.Gly52Asp | PM1, PM2, PP3 | VUS | 0.527 | 0.050 | 0.591 | c.2119_2120delGT |
| BEST1 | Best Vitelliform Macular Dystrophy (Dominant) | NM_001139443.1 | chr11 | g.61955783 | c.133C>G | p.Arg45Gly | PM2, PP3, PP4(S) | Likely Pathogenic | 0.966 | 0.699 | 0.945 | / |
| BEST1 | Best Vitelliform Macular Dystrophy (Dominant) | NM_001139443.1 | chr11 | g.61955157 | c.23A>G | p.Tyr8Cys | PM2, PP3, PP4(S) | Likely Pathogenic | 0.976 | 0.710 | 0.917 | / |
| CLRN1 | USH Type III | NM_001195794.1 | chr3 | g.150928039 | c.635A>G | p.His212Arg | PM2, PM3, PP1, PP3 | Likely Pathogenic | 0.573 | 0.055 | 0.799 | c.118T>G |
| CNGA3 | Achromatopsia | NM_001298.2 | chr2 | g.98391904 | c.607T>C | p.Trp203Arg | PM2, PP3 | VUS | 0.960 | 0.316 | 0.950 | c.1694C>T |
| MYO7A | USH Type I | NM_000260.3 | chr11 | g.77192227 | c.4101C>G | p.Ile1367Met | PM2, PP3 | VUS | 0.542 | 0.220 | 0.672 | c.2904G>T |
| PEX7 | Uncategorised Syndrome | NM_000288.3 | chr6 | g.136822742 | c.77C>T | p.Pro26Leu | PM2, PP3, PP4 | VUS | 0.962 | 0.947 | 0.864 | c.875T>A |
| PRPF31 | Retinitis Pigmentosa (Simplex) | NM_015629.3 | chr19 | g.54124558 | c.757G>A | p.Gly253Arg | PM2, PP3 | VUS | 0.946 | 0.911 | 0.957 | / |
| PRPH2 | Retinitis Pigmentosa (Dominant) | NM_000322.4 | chr6 | g.42721878 | c.457A>G | p.Lys153Glu | PM2, PP3 | VUS | 0.628 | 0.180 | 0.864 | / |
| PRPH2 | Retinitis Pigmentosa (Dominant) | NM_000322.4 | chr6 | g.42721871 | c.464C>T | p.Thr155Ile | PM2, PP3 | VUS | 0.518 | 0.100 | 0.649 | / |
| RDH5 | Fundus Albipunctatus | NM_001199771.1 | chr12 | g.55721276 | c.92T>C | p.Val31Ala | PM2, PP3 | VUS | 0.670 | 0.143 | 0.737 | c.712G>T |
| USH2A | USH Type II | NM_206933.2 | chr1 | g.216247097 | c.2297G>A | p.Cys766Tyr | PM2, PM3, PM5, PP3 | Likely Pathogenic | 0.945 | 0.669 | 0.952 | c.2299delG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whelan, L.; Dockery, A.; Wynne, N.; Zhu, J.; Stephenson, K.; Silvestri, G.; Turner, J.; O’Byrne, J.J.; Carrigan, M.; Humphries, P.; et al. Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland. Genes 2020, 11, 105. https://doi.org/10.3390/genes11010105
Whelan L, Dockery A, Wynne N, Zhu J, Stephenson K, Silvestri G, Turner J, O’Byrne JJ, Carrigan M, Humphries P, et al. Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland. Genes. 2020; 11(1):105. https://doi.org/10.3390/genes11010105
Chicago/Turabian StyleWhelan, Laura, Adrian Dockery, Niamh Wynne, Julia Zhu, Kirk Stephenson, Giuliana Silvestri, Jacqueline Turner, James J. O’Byrne, Matthew Carrigan, Peter Humphries, and et al. 2020. "Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland" Genes 11, no. 1: 105. https://doi.org/10.3390/genes11010105
APA StyleWhelan, L., Dockery, A., Wynne, N., Zhu, J., Stephenson, K., Silvestri, G., Turner, J., O’Byrne, J. J., Carrigan, M., Humphries, P., Keegan, D., Kenna, P. F., & Farrar, G. J. (2020). Findings from a Genotyping Study of over 1000 People with Inherited Retinal Disorders in Ireland. Genes, 11(1), 105. https://doi.org/10.3390/genes11010105