1. Introduction
Marek’s disease (MD) is a T cell lymphoma and a strictly cell-associated disease induced by the highly oncogenic α-herpesvirus II disease virus [
1], which has a complex life with four main phases [
2,
3]: an early cytolytic phase at 2–7 days post infection (dpi), a latent phase around 7–10 dpi, a late cytolytic phase with the presence of tumors that is triggered between 14 and 21 dpi, and a final proliferation phase after 28 dpi. During the first cytolytic phase, Marek’s disease virus (MDV) first uses B cells as targets for its replication before targeting activated CD4
+ T cells to enable a persistent latent infection [
4,
5,
6]. MDV then uses T cells as the main target during the later phase, especially CD4
+ T cells [
7] during the cytolytic phase with tumors at 21 dpi, which were used in this study. CD4 is a co-receptor that facilitates T cell receptor (TCR) communication with an antigen-presenting cell. MD is a commercially important neoplastic disease in chickens and also the main chronic infectious disease threatening the poultry industry. Enhancing genetic resistance to MD in poultry is an important long-term goal in controlling MD. To optimally implement this control strategy through marker assisted selection (MAS) and to understand the etiology and mechanisms of MD, it is necessary to identify more specific alleles and genes with respect to MD latency, although alleles at the well-studied major histocompatibility complex (MHC) locus are already known to be involved in genetic resistance to MD.
The comprehensive identification of genes underlying phenotypic variation of complex traits, especially disease resistance, remains one of the greatest challenges in biology, despite having genome sequences and powerful tools. Screening for allele-specific expression (ASE) [
8,
9,
10,
11] is an effective approach to identifying regulatory variation responsible for differences in transcript abundance in genes. ASE refers to unequal expression of multiple alleles of a gene, and the extreme case of ASE is monoallelic expression, where only one of the alleles is expressed while the other is completely inactive. The genes with single nucleotide polymorphisms (SNPs) exhibiting ASE will provide a strong foundation for investigating the genetic mechanisms of complex traits, such as MDV infection. Cheng et al. [
12] found that SNPs in ASE genes account for more than 83% of the additive genetic variation of genetic resistance to MD, demonstrating that most of the genes exhibiting ASE are strong candidates for studying MD resistance. MacEachern et al. [
13] suggested that ASE can be used to identify genes with cis-regulatory elements that respond to MDV infection. Perumbakkam et al. [
14] considered that the TLR and JAK/STAT signaling pathways may be responding to MDV infection through the genes exhibiting ASE.
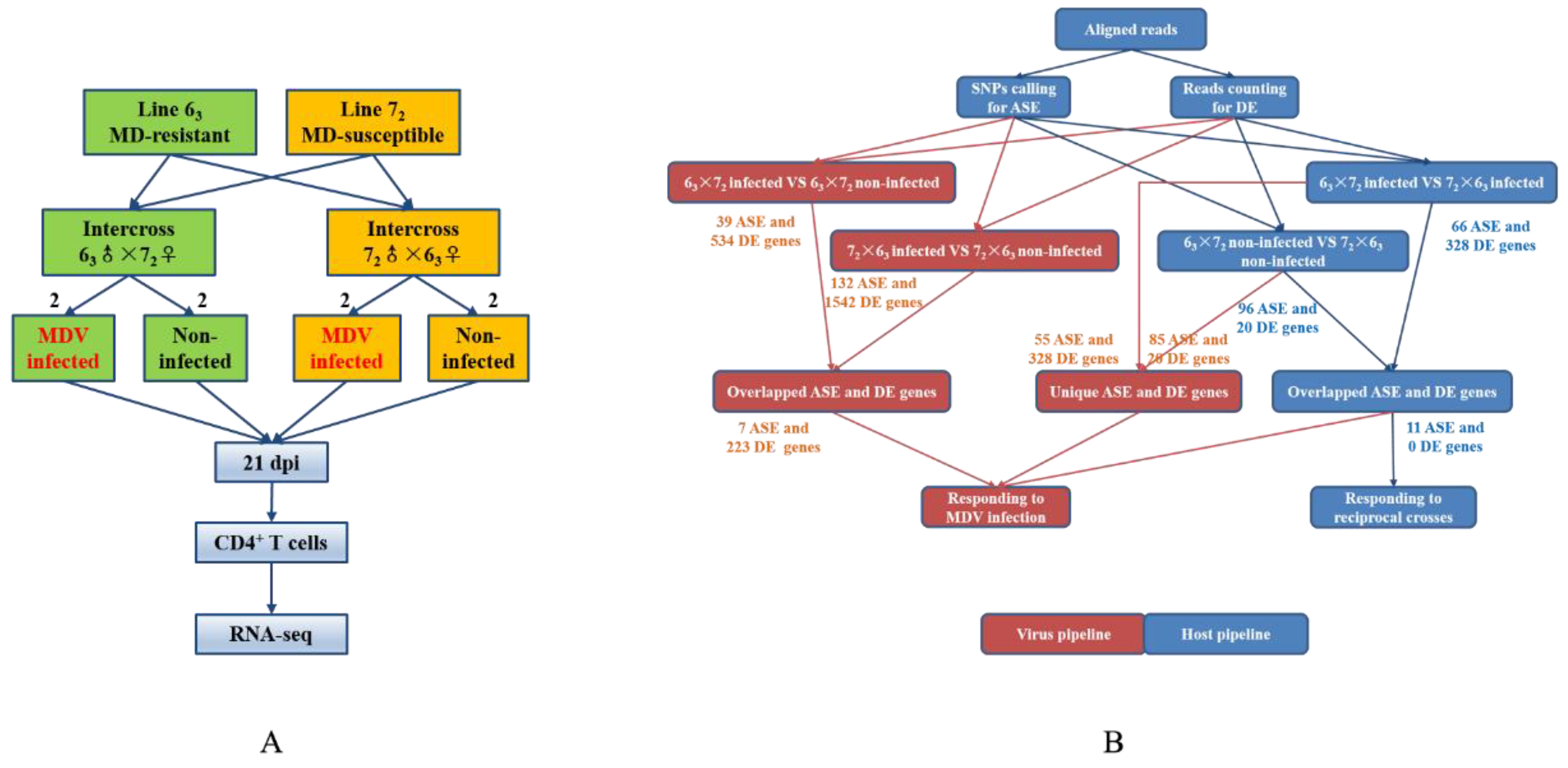
In the present study, transcriptomic data in CD4+ T cells isolated from MDV-infected and non-infected groups of two reciprocal crosses were analyzed by high throughput sequencing (RNA-seq) to identify candidate genes of MD genetic resistance. Two F1 reciprocal crosses were used to compare the differences, not only between infected and non-infected individuals, but also between two reciprocal crosses. Key SNPs, genes, and pathways related to MD resistance or MD susceptibility were identified by ASE and differential expression (DE) analyses. We used both CD4+ T cells and tumors for the validation of genes exhibiting ASE.
4. Discussion
MD, a complicated tumor disease, has been used as a model to study human tumors [
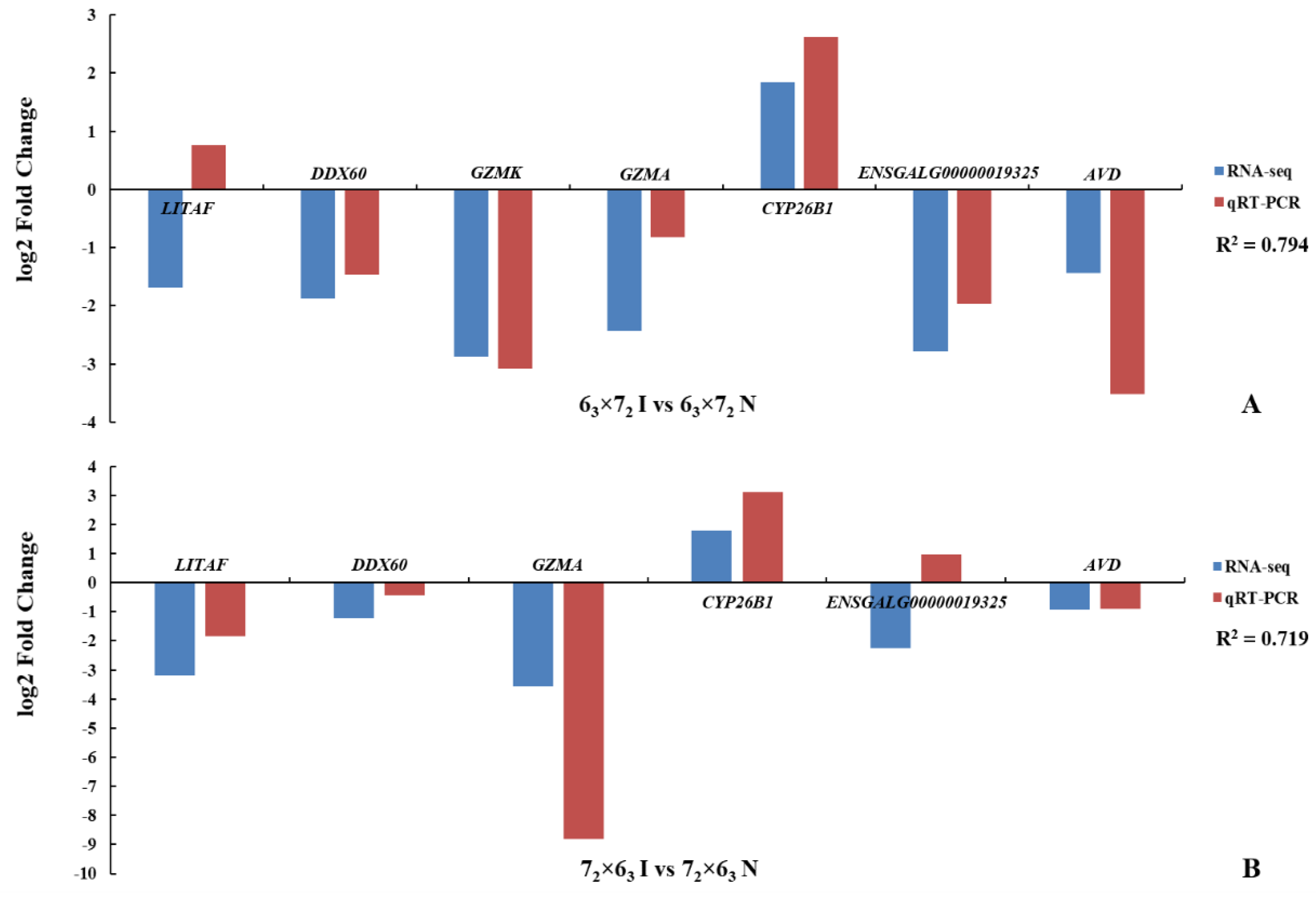
31]. The genetic mechanism underlying MD resistance and MD susceptibility is likely to be complex and remains incompletely understood. Thus, understanding the genetic basis of MD resistance or MD susceptibility for poultry is important to provide crucial clues for human diseases. In the present study, based on the high throughput sequencing platform, some bioinformatics analyses, such as ASE, DE, GO, and pathway analysis, were used to identify SNPs, genes, and enriched pathways from infected and non-infected birds. Six genes exhibiting ASE and six pathways were selected as the likely candidate factors that induce MD resistance. To minimize transcriptional variations and take full advantage of identical genetic backgrounds in inbred lines, we paired birds by reciprocal crossing and MDV infection and tested the differences. We not only identified genes most likely related to MD resistance, but also revealed the effects of MDV infection on the host.
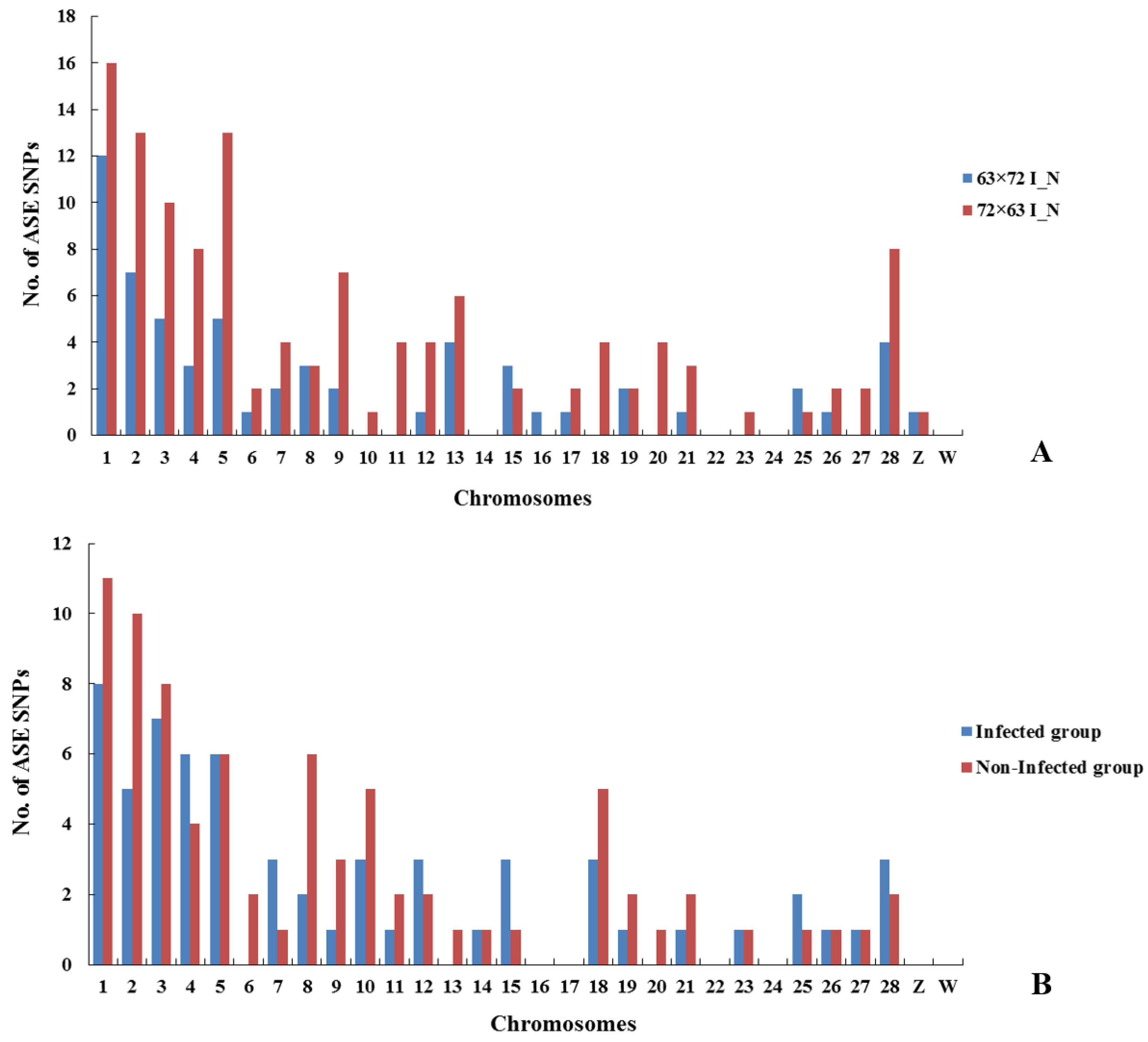
ASE is a powerful technique used to measure the expression of each allele through studying SNP within an RNA sample [
32,
33,
34,
35]. The F
1 data used here are an ideal test case for our approach. In this study, we hypothesized that a gene with allelic inequality or imbalance is the key genetic factor causing MDV susceptibility. Since variation in gene expression is thought to be a major factor affecting phenotypic variation, genes with ASE SNPs provide candidates and markers that may account for the complex trait of interest. Although the ASE SNPs identified here could not be considered to explain much more additive genetic variation of the genetic resistance to MD because fewer ASE SNPs and genes were obtained from RNA-seq data, our findings are still of interest in view of the strict experimental design and analysis methods. In this experiment, we successfully aligned RNA-seq reads to the reference chicken genome and identified high quality SNPs in infected and non-infected chickens. The significant SNPs exhibiting ASE in response to MDV infection were distributed on autosomes 1 to 28 and Z and W chromosomes of the chicken genome, with the number of SNPs for each chromosome roughly proportional to the size of each chromosome in the chicken genome assembly. To locate these SNPs on the chicken genome with respect to genes and classify them based on function, VEP was used. In the 6
3 × 7
2 I_N group, the most SNPs (
n = 19, 31.1%) were classified as exonic; however, the largest number of SNPs (
n = 52, 32.7%) was classified as intronic in the 7
2 × 6
3 I_N group. The largest number of SNPs was classified as intronic (
n = 26, 34.7%) and located downstream of a gene (
n =23, 20.5%) in infected and non-infected groups, respectively. Interestingly, for both ASE and DE results, more SNPs and DEGs were identified in the 7
2 × 6
3 I_N group than in the 6
3 × 7
2 I_N group, which may be because of the different MD incidences (%) of both reciprocal crosses. Seven overlapped ASE genes were identified in intercrosses 6
3 × 7
2 and 7
2 × 6
3 in response to MDV infection. We also identified 11 overlapped ASE genes in infected and non-infected groups, and these common genes are likely induced by MD viral infection in the host.
MCL1 was the only gene identified in both overlapped groups, which could be firstly selected as a key gene in the response to MDV infection. Other ASE genes, including
CD5,
CD28,
CD44,
TCF1, and
IRF-4, are also important genes related to T cells, viruses, and tumors. DE is another powerful technique that could further explain the results of ASE. Many more overlapped genes were identified in DE than in ASE analysis in response to MDV infection, including
MCL1.
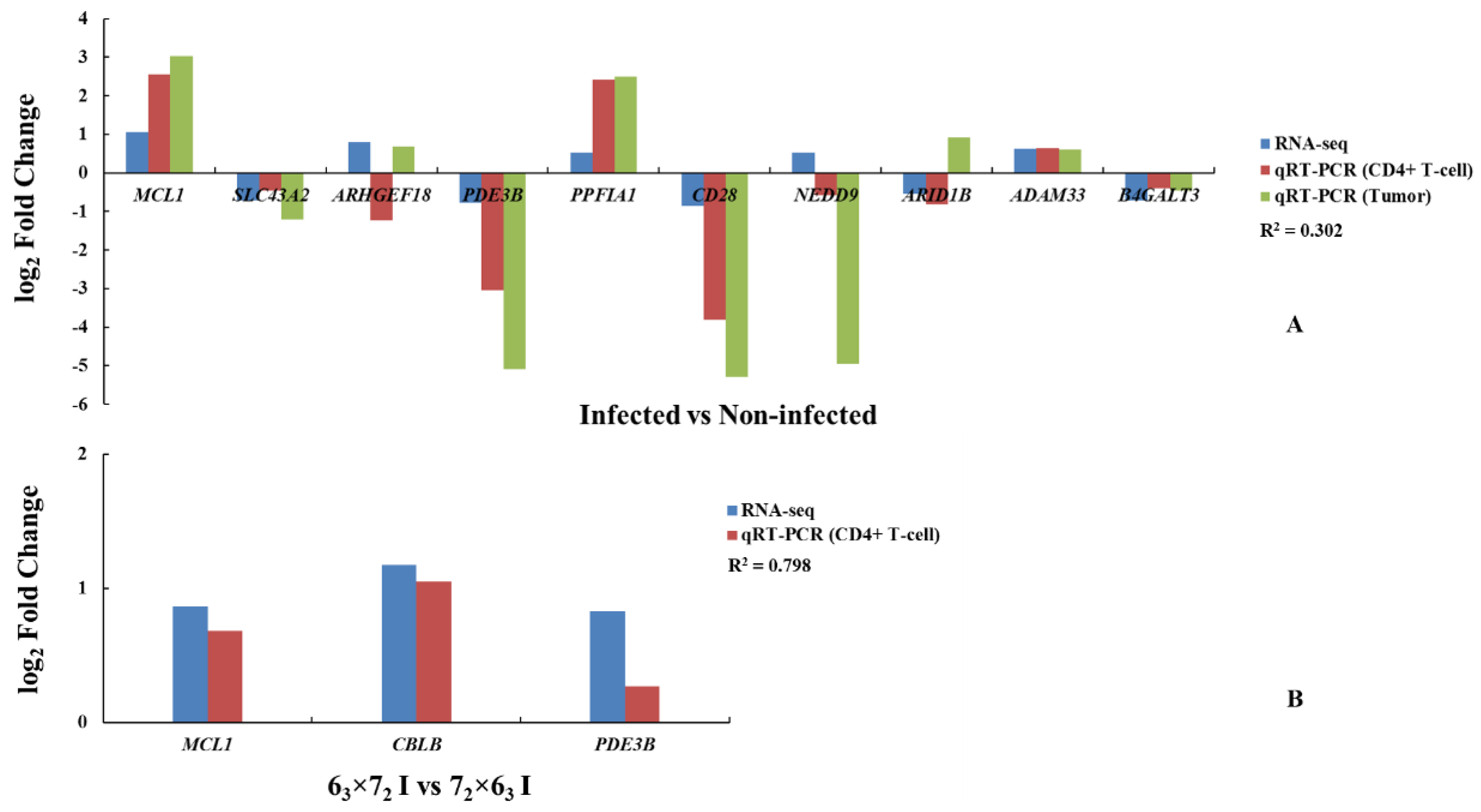
We compared the results of the DEGs to the ASE gene analysis to identify four overlapped genes:
MCL1,
SLC43A2,
PDE3B, and
ADAM33. As a tumor disease, gene functional validation was conducted not only in CD4
+ T cells but also in tumor tissues, which could provide powerful evidence of key genes responding to MDV resistance. Eight genes exhibiting ASE, including these four genes, showed significant differences in both CD4
+ T cells and tumors of the comparison groups, and were completely consistent with the RNA-seq results. Therefore, these four genes could be selected as potential candidate genes to study MD resistance or MD susceptibility, of which
MCL1, as a member of the
BCL2 family, plays a role in cell proliferation, differentiation, tumorigenesis, and apoptosis [
36,
37,
38,
39]. It is also associated with tumor, cancer, and CD4
+ and CD8
+ T cells [
40,
41,
42,
43]. In this study, we found an ASE SNP in this
MCL1 gene, and the expression of this gene is significantly higher in infected birds and tumors. Thus, this SNP may increase the gene expression of
MCL1 after MDV infection. This gene was identified both as a virus-related gene and a host-related gene, so it could be a critical gene connecting MDV to the host.
Chicken MHC plays an important role in the determination of resistance to MDV [
1]. The GGA16 contains three loci, the B locus, Y (or Rfp-Y) locus, and nucleolar-organizing region (NOR), and only one ASE SNP was found to be associated with a gene that encodes for the class I alpha chain of the Rfp-Y loci [
14]. In this study, no SNPs in the related loci were identified, and this could be attributed to the incomplete sequence information available for the GGA16 loci in the chicken genome. However, in the DE analysis, two MHC class II (MHCII)-associated genes,
BLB1 [
44,
45,
46] and
DMB2 [
47,
48,
49], were found to be significantly different between infected and non-infected birds, which are related to T cell immune responses, and their expressions are altered in cancers and tumors.
Due to the complexity of MD, it may not be a single-gene-controlled trait. Thus, we subsequently performed pathway analyses to investigate the specific gene sets involved in signaling cascades. DAVID analysis is usually a simple and useful method that is widely used for many gene set enrichment analyses. IPA is a powerful and useful commercial gene set enrichment analysis tool. In this study, based on the DAVID and IPA results, we focused on several important pathways that may be related to MDV infection. T cell activation [
50] was identified by DAVID from the PANTHER pathway dataset. Activation of CD4
+ T cells occurs through the simultaneous engagement of the T cell receptor and a co-stimulatory molecule on the T cell via the MHCII peptide and co-stimulatory molecules on the antigen-presenting cell (APC). This signaling pathway downstream from co-stimulatory molecules engages the PI3K/AKT molecules, which was identified in both DAVID and IPA analysis. The PI3K/AKT-mTOR signaling pathway is one of the three major signaling pathways critical in tumor progression. This classic route consists of two signaling pathways, PI3K/AKT and mTOR, which are important in regulating the cell cycle and directly related to cellular proliferation, tumors, and cancer. AKT is activated downstream of PI3K and then activates mTOR [
51]. In many cancers, this pathway is overactive, thus reducing apoptosis and allowing proliferation. This pathway is related to the T cell activation pathway mentioned above and the key MD-related gene
MCL1, identified in this study, is also involved in this pathway. This pathway may play a role in MD resistance or MD susceptibility. The TCR signaling pathway was the top canonical pathway identified by IPA. TCR is a molecule found on the surfaces of T cells or T lymphocytes, and is responsible for recognizing fragments of antigens as peptides bound to MHC molecules. When the TCR engages with an antigenic peptide and MHC (peptide/MHC), the T lymphocyte is activated through signal transduction, which is a series of biochemical events mediated by associated enzymes, co-receptors (like CD4), specialized adaptor molecules, and activated or released transcription factors [
52]. MDV first uses B cells as targets for its replication before targeting activated CD4
+ T cells. Thus, the BCR signaling pathway is critical for MDV infection. BCR is a transmembrane receptor protein located on the outer surface of B cells that has two crucial functions: signal transduction, involving changes in receptor oligomerization, and mediating internalization for subsequent processing of the antigen and presentation of peptides to helper T cells. BCR functions are required for normal antibody production, so defects in BCR signal transduction may lead to immunodeficiency [
53]. The ERK/MAPK signaling pathway is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell. It consists of many proteins, including mitogen-activated protein kinase (MAPK), which is necessary for the development of cancers [
54]. This pathway is one of the most extensively studied pathways involved in tumorigenesis [
55] and has been found to be critical in previous MD studies by Yan et al. [
56] and Subramaniam et al. [
57,
58]. Overall, our analyses indicate that the genes and pathways described here are worthy of further studies on MD.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}