Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Growth Conditions
2.2. Patients and Ethics Statements
2.3. RNA Purification and Reverse Transcription
2.4. Rapid Amplification of cDNA 3′ Ends (3′ RACE)
2.5. PCR and Sequencing
2.6. Statistical Analysis
3. Results
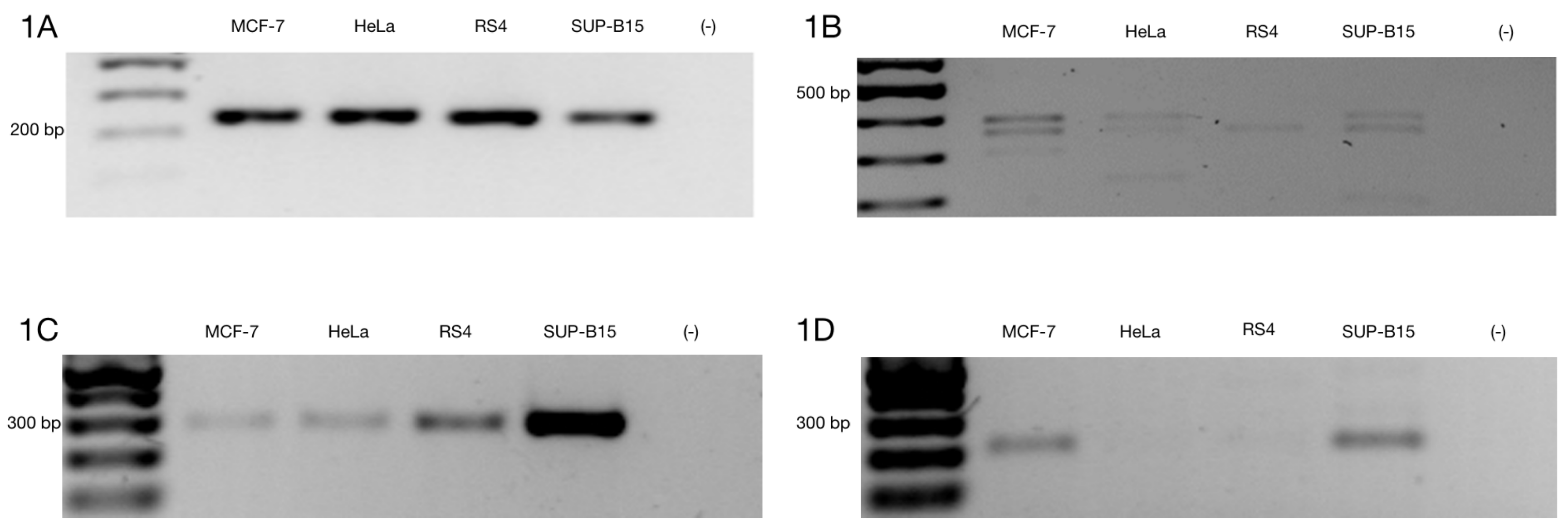
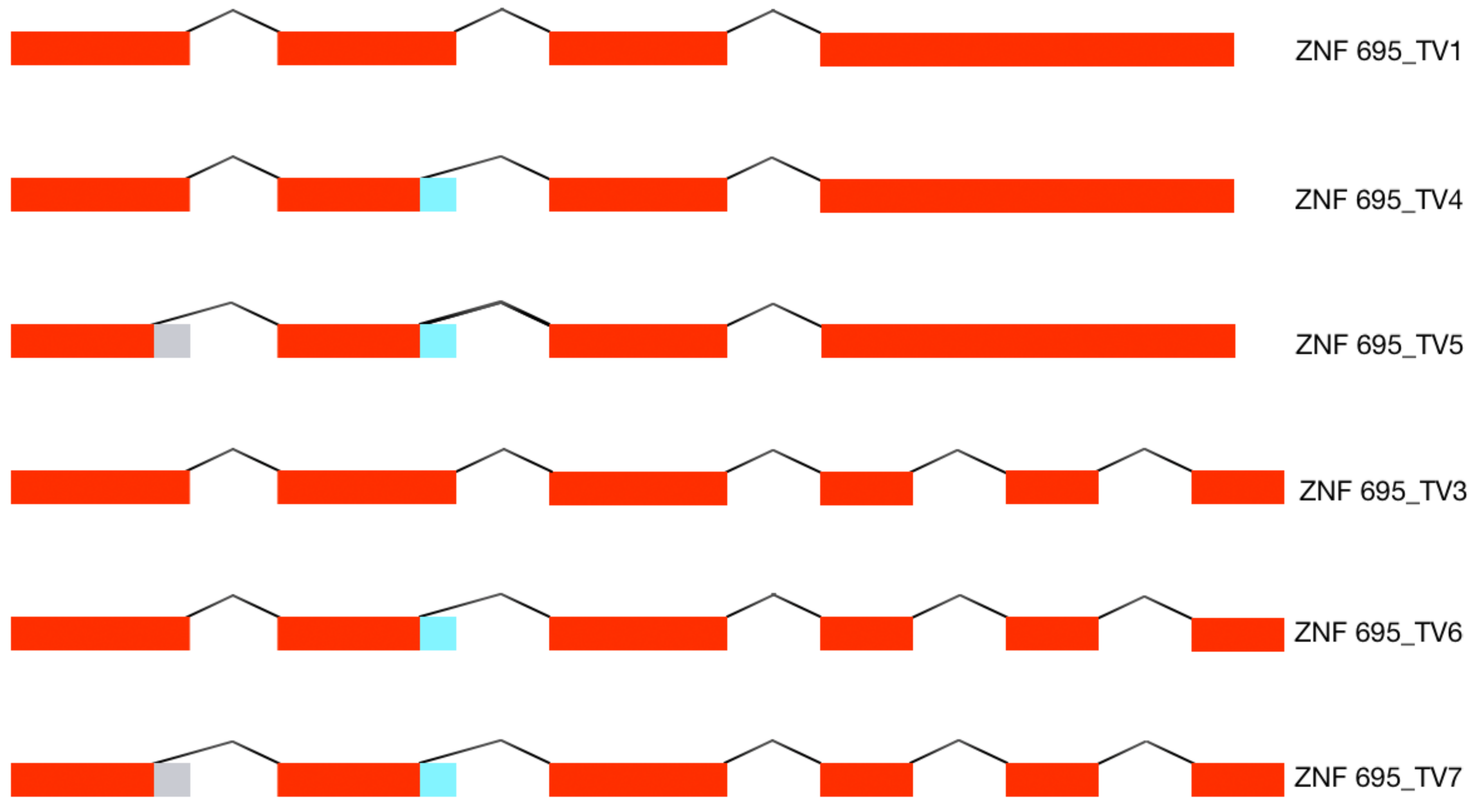
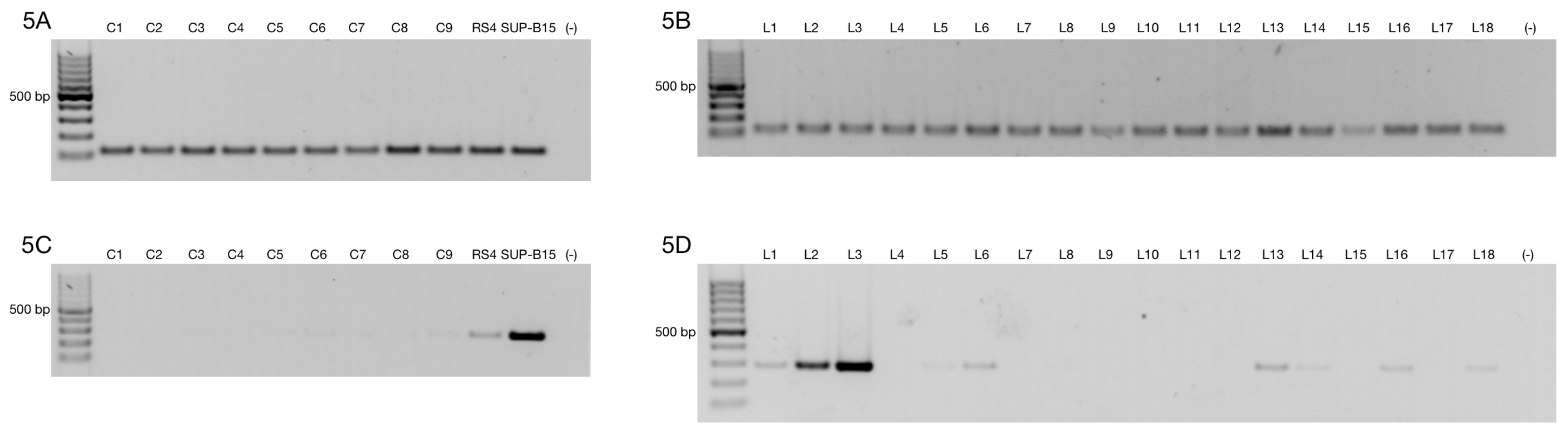
3.1. Alternative ZNF695 Transcript Variants Are Expressed in Cancer Cell Lines
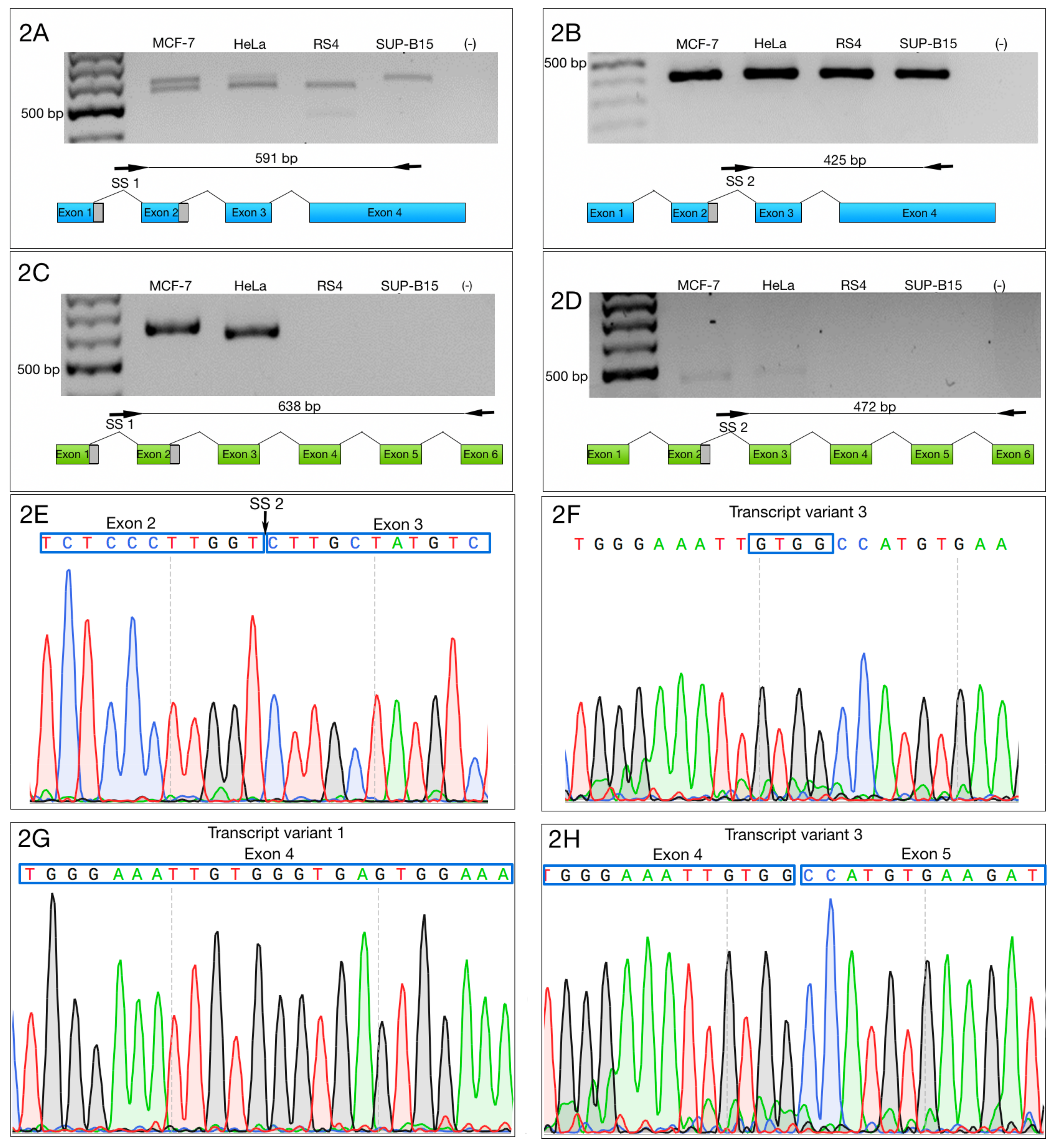
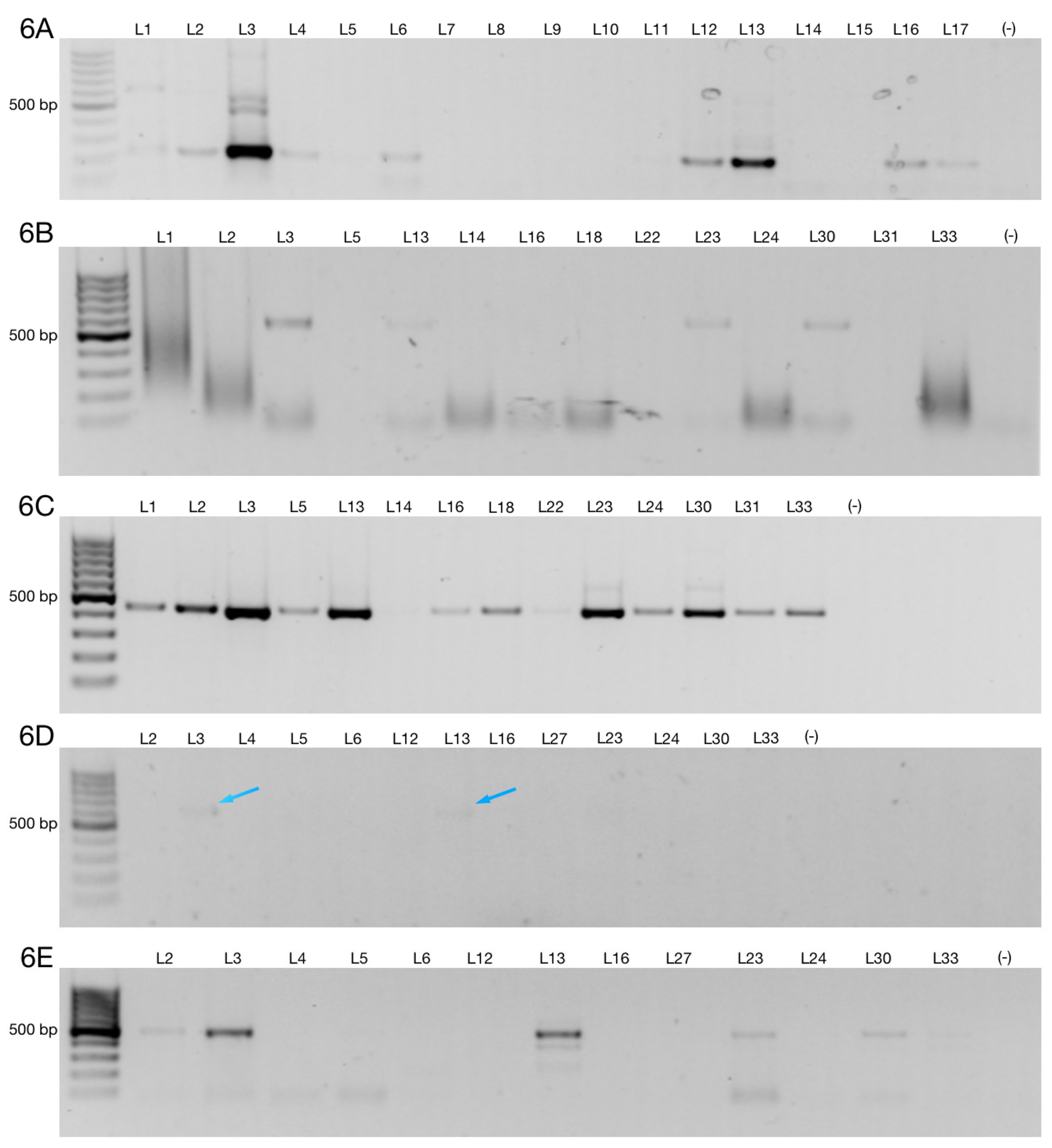
3.2. Alternative 3′ Ends in ZNF695 Transcript Variants
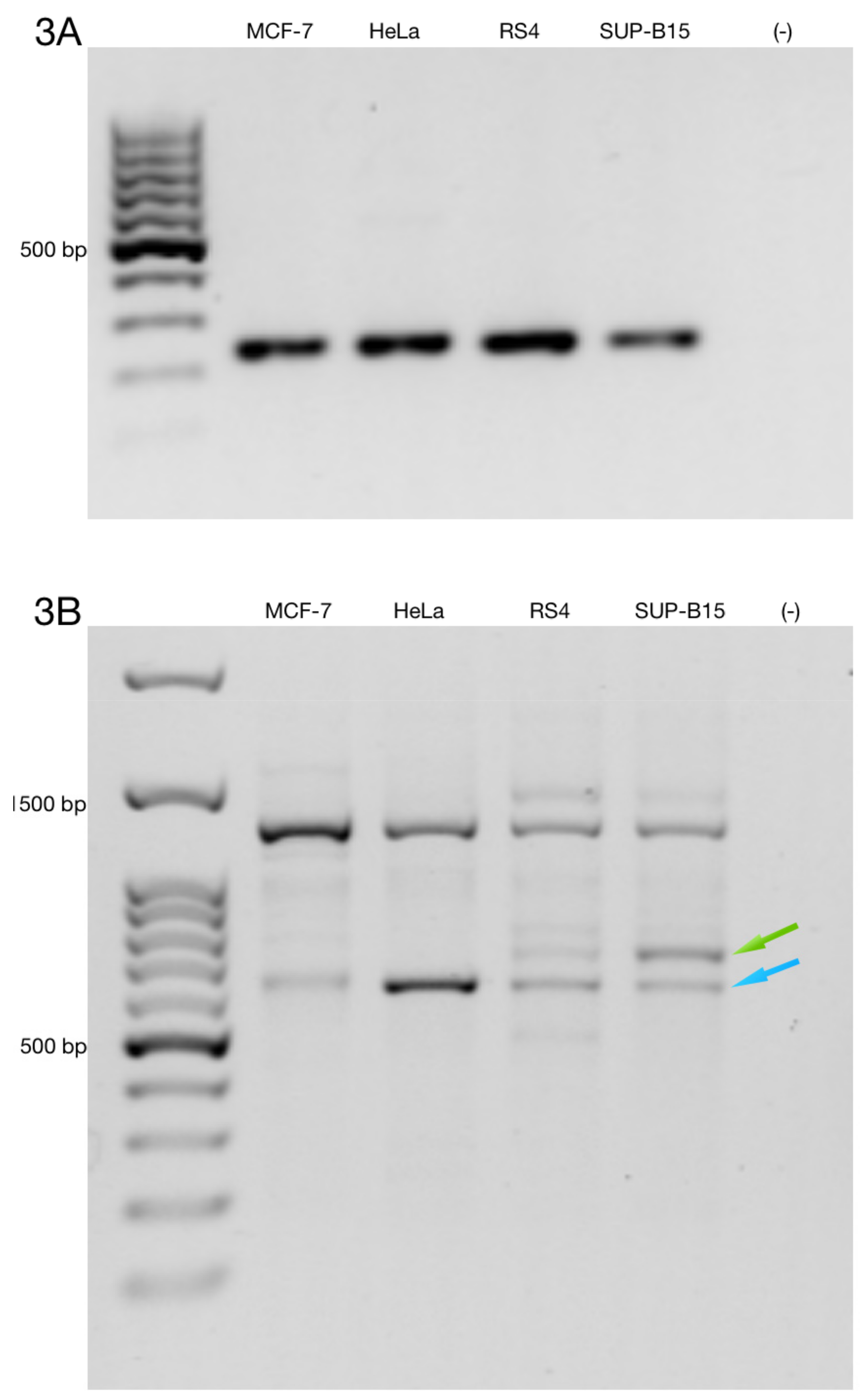
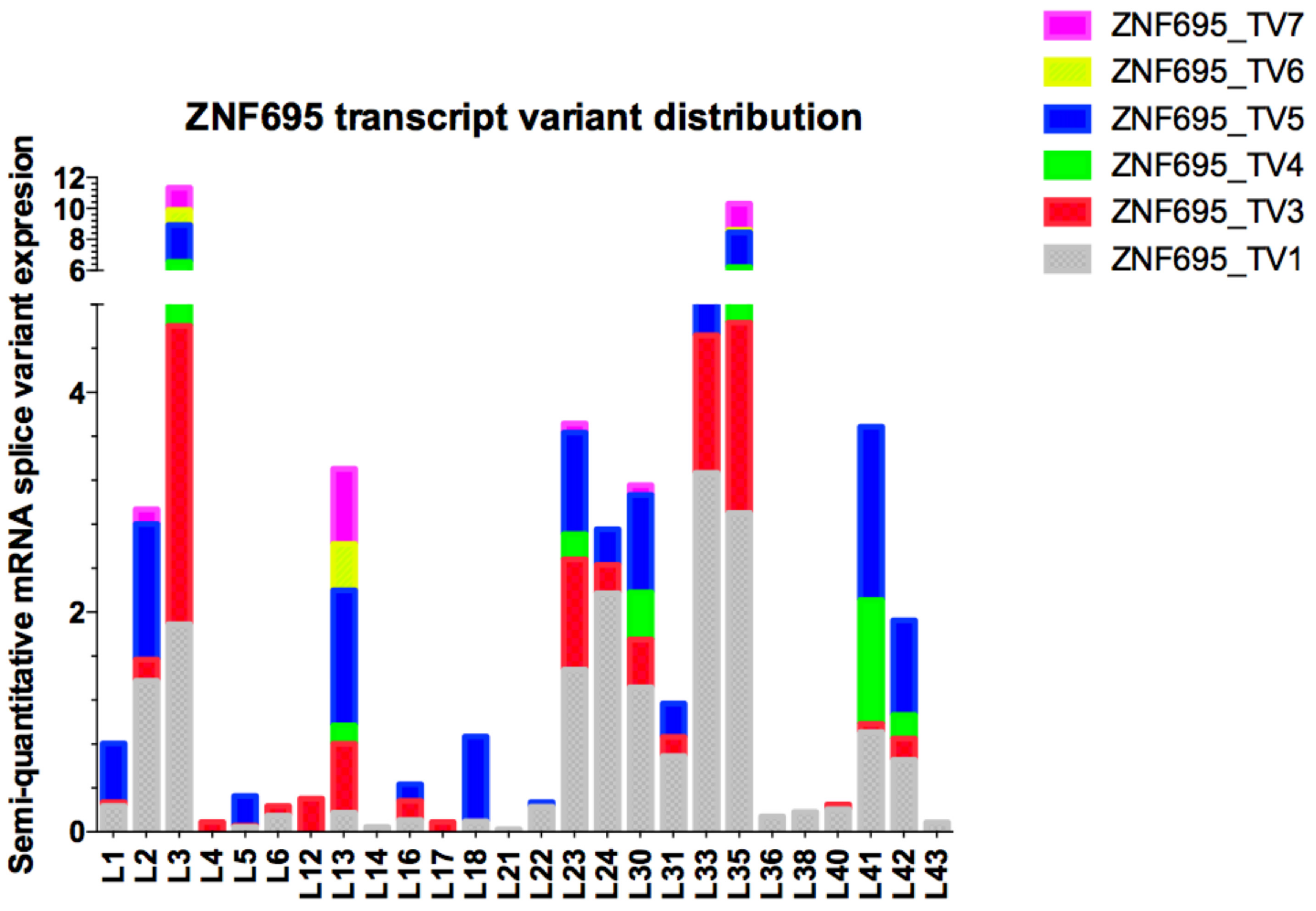
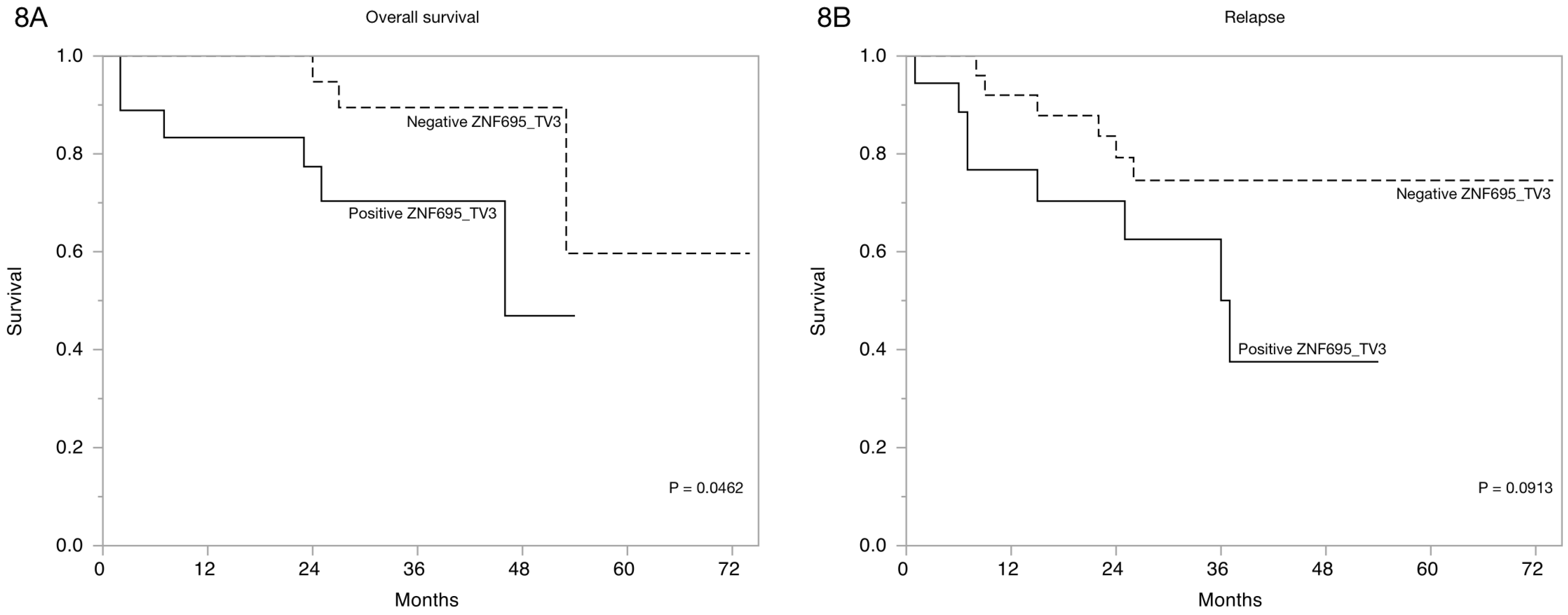
3.3. Alternative ZNF695 Transcript Variants Are Expressed in B-ALL
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Almamun, M.; Kholod, O.; Stuckel, A.J.; Levinson, B.T.; Johnson, N.T.; Arthur, G.L.; Davis, J.W.; Taylor, K.H. Inferring a role for methylation of intergenic DNA in the regulation of genes aberrantly expressed in precursor B-cell acute lymphoblastic leukemia. Leuk. Lymphoma 2017, 58, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Pritchard-Jones, K.; Pieters, R.; Reaman, G.H.; Hjorth, L.; Downie, P.; Calaminus, G.; Naafs-Wilstra, M.C.; Steliarova-Foucher, E. Sustaining innovation and improvement in the treatment of childhood cancer: Lessons from high-income countries. Lancet Oncol. 2013, 14, e95–e103. [Google Scholar] [CrossRef]
- Magrath, I.; Steliarova-Foucher, E.; Epelman, S.; Ribeiro, R.C.; Harif, M.; Li, C.K.; Kebudi, R.; Macfarlane, S.D.; Howard, S.C. Paediatric cancer in low-income and middle-income countries. Lancet Oncol. 2013, 14, e104–e116. [Google Scholar] [CrossRef]
- Deng, Y.; Liu, B.; Fan, X.; Wang, Y.; Tang, M.; Mo, X.; Li, Y.; Ying, Z.; Wan, Y.; Luo, N.; et al. ZNF552, a novel human KRAB/C2H2 zinc finger protein, inhibits AP-1- and SRE-mediated transcriptional activity. BMB Rep. 2010, 43, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Nalabolu, S.R.; Aster, J.C.; Ma, J.; Abruzzo, L.; Jaffe, E.S.; Stone, R.; Weissman, S.M.; Hudson, T.J.; Fletcher, J.A. FGFR1 is fused with a novel zinc-finger gene, ZNF198, in the t(8;13) leukaemia/lymphoma syndrome. Nat. Genet. 1998, 18, 84–87. [Google Scholar] [CrossRef]
- O’Geen, H.; Squazzo, S.L.; Iyengar, S.; Blahnik, K.; Rinn, J.L.; Chang, H.Y.; Green, R.; Farnham, P.J. Genome-wide analysis of KAP1 binding suggests autoregulation of KRAB-ZNFs. PloS Genet. 2007, 3, e89. [Google Scholar] [CrossRef]
- Vogel, M.J.; Guelen, L.; de Wit, E.; Peric-Hupkes, D.; Loden, M.; Talhout, W.; Feenstra, M.; Abbas, B.; Classen, A.K.; van Steensel, B. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 2006, 16, 1493–1504. [Google Scholar] [CrossRef]
- Blahnik, K.R.; Dou, L.; Echipare, L.; Iyengar, S.; O’Geen, H.; Sanchez, E.; Zhao, Y.; Marra, M.A.; Hirst, M.; Costello, J.F.; et al. Characterization of the contradictory chromatin signatures at the 3′ exons of zinc finger genes. PLoS ONE 2011, 6, e17121. [Google Scholar] [CrossRef]
- Grondin, B.; Cote, F.; Bazinet, M.; Vincent, M.; Aubry, M. Direct interaction of the KRAB/Cys2-His2 zinc finger protein ZNF74 with a hyperphosphorylated form of the RNA polymerase II largest subunit. J. Biol. Chem. 1997, 272, 27877–27885. [Google Scholar] [CrossRef]
- Cote, F.; Boisvert, F.M.; Grondin, B.; Bazinet, M.; Goodyer, C.G.; Bazett-Jones, D.P.; Aubry, M. Alternative promoter usage and splicing of ZNF74 multifinger gene produce protein isoforms with a different repressor activity and nuclear partitioning. DNA Cell Biol. 2001, 20, 159–173. [Google Scholar] [CrossRef]
- Takashima, H.; Nishio, H.; Wakao, H.; Nishio, M.; Koizumi, K.; Oda, A.; Koike, T.; Sawada, K. Molecular cloning and characterization of a KRAB-containing zinc finger protein, ZNF317, and its isoforms. Biochem. Biophys. Res. Commun. 2001, 288, 771–779. [Google Scholar] [CrossRef]
- Urrutia, R. KRAB-containing zinc-finger repressor proteins. Genome Biol. 2003, 4, 231. [Google Scholar] [CrossRef]
- Resch, A.; Xing, Y.; Modrek, B.; Gorlick, M.; Riley, R.; Lee, C. Assessing the impact of alternative splicing on domain interactions in the human proteome. J. Proteome Res. 2004, 3, 76–83. [Google Scholar] [CrossRef]
- Shao, H.; Zhu, C.; Zhao, Z.; Guo, M.; Qiu, H.; Liu, H.; Wang, D.; Xue, L.; Gao, L.; Sun, C.; et al. KRAB-containing zinc finger gene ZNF268 encodes multiple alternatively spliced isoforms that contain transcription regulatory domains. Int. J. Mol. Med. 2006, 18, 457–463. [Google Scholar] [CrossRef][Green Version]
- Chun, J.N.; Song, I.S.; Kang, D.H.; Song, H.J.; Kim, H.I.; Seo, J.; Lee, K.J.; Kim, J.; Kang, S.W. A splice variant of the C(2)H(2)-type zinc finger protein, ZNF268s, regulates NF-kappaB activation by TNF-α. Mol. Cells 2008, 26, 175–180. [Google Scholar]
- Kahns, S.; Losson, R.; Nielsen, A.L. Nizp1 zinc finger protein localization is determined by SCAN-domain inclusion regulated through alternative splicing. Biochim. Biophys. Acta 2010, 1799, 539–545. [Google Scholar] [CrossRef]
- Margolin, J.F.; Friedman, J.R.; Meyer, W.K.; Vissing, H.; Thiesen, H.J.; Rauscher, F.J., 3rd. Kruppel-associated boxes are potent transcriptional repression domains. Proc. Natl. Acad. Sci. USA 1994, 91, 4509–4513. [Google Scholar] [CrossRef]
- Vissing, H.; Meyer, W.K.; Aagaard, L.; Tommerup, N.; Thiesen, H.J. Repression of transcriptional activity by heterologous KRAB domains present in zinc finger proteins. FEBS Lett. 1995, 369, 153–157. [Google Scholar] [CrossRef]
- Witzgall, R.; O’Leary, E.; Leaf, A.; Onaldi, D.; Bonventre, J.V. The Kruppel-associated box-A (KRAB-A) domain of zinc finger proteins mediates transcriptional repression. Proc. Natl. Acad. Sci. USA 1994, 91, 4514–4518. [Google Scholar] [CrossRef]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J., 3rd. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef]
- Moosmann, P.; Georgiev, O.; Le Douarin, B.; Bourquin, J.P.; Schaffner, W. Transcriptional repression by RING finger protein TIF1 β that interacts with the KRAB repressor domain of KOX1. Nucleic Acids Res. 1996, 24, 4859–4867. [Google Scholar] [CrossRef]
- Underhill, C.; Qutob, M.S.; Yee, S.P.; Torchia, J. A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 2000, 275, 40463–40470. [Google Scholar] [CrossRef]
- Abrink, M.; Ortiz, J.A.; Mark, C.; Sanchez, C.; Looman, C.; Hellman, L.; Chambon, P.; Losson, R. Conserved interaction between distinct Kruppel-associated box domains and the transcriptional intermediary factor 1 β. Proc. Natl. Acad. Sci. USA 2001, 98, 1422–1426. [Google Scholar] [CrossRef]
- Lorenz, P.; Koczan, D.; Thiesen, H.J. Transcriptional repression mediated by the KRAB domain of the human C2H2 zinc finger protein Kox1/ZNF10 does not require histone deacetylation. Biol. Chem. 2001, 382, 637–644. [Google Scholar] [CrossRef]
- Bellefroid, E.J.; Poncelet, D.A.; Lecocq, P.J.; Revelant, O.; Martial, J.A. The evolutionarily conserved Kruppel-associated box domain defines a subfamily of eukaryotic multifingered proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 3608–3612. [Google Scholar] [CrossRef]
- Thiesen, H.J.; Bellefroid, E.; Revelant, O.; Martial, J.A. Conserved KRAB protein domain identified upstream from the zinc finger region of Kox 8. Nucleic Acids Res. 1991, 19, 3996. [Google Scholar] [CrossRef][Green Version]
- Lovering, R.; Trowsdale, J. A gene encoding 22 highly related zinc fingers is expressed in lymphoid cell lines. Nucleic Acids Res. 1991, 19, 2921–2928. [Google Scholar] [CrossRef][Green Version]
- Rosati, M.; Marino, M.; Franze, A.; Tramontano, A.; Grimaldi, G. Members of the zinc finger protein gene family sharing a conserved N-terminal module. Nucleic Acids Res. 1991, 19, 5661–5667. [Google Scholar] [CrossRef][Green Version]
- Sugnet, C.W.; Kent, W.J.; Ares, M., Jr.; Haussler, D. Transcriptome and genome conservation of alternative splicing events in humans and mice. Pac. Symp. Biocomput. Pac. Symp. Biocomput. 2004, 66–77. [Google Scholar] [CrossRef]
- Manley, J.L.; Tacke, R. SR proteins and splicing control. Genes Dev. 1996, 10, 1569–1579. [Google Scholar] [CrossRef]
- Caceres, J.F.; Kornblihtt, A.R. Alternative splicing: Multiple control mechanisms and involvement in human disease. Trends Genet. TIG 2002, 18, 186–193. [Google Scholar] [CrossRef]
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243. [Google Scholar] [CrossRef]
- Cartegni, L.; Chew, S.L.; Krainer, A.R. Listening to silence and understanding nonsense: Exonic mutations that affect splicing. Nat. Rev. Genet. 2002, 3, 285–298. [Google Scholar] [CrossRef]
- Cartegni, L.; Krainer, A.R. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat. Genet. 2002, 30, 377–384. [Google Scholar] [CrossRef]
- Wagner, E.J.; Garcia-Blanco, M.A. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 2001, 21, 3281–3288. [Google Scholar] [CrossRef]
- Faustino, N.A.; Cooper, T.A. Pre-mRNA splicing and human disease. Genes Dev. 2003, 17, 419–437. [Google Scholar] [CrossRef]
- Lareau, L.F.; Green, R.E.; Bhatnagar, R.S.; Brenner, S.E. The evolving roles of alternative splicing. Curr. Opin. Struct. Biol. 2004, 14, 273–282. [Google Scholar] [CrossRef]
- Sveen, A.; Kilpinen, S.; Ruusulehto, A.; Lothe, R.A.; Skotheim, R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene 2016, 35, 2413–2427. [Google Scholar] [CrossRef]
- Philips, A.V.; Cooper, T.A. RNA processing and human disease. Cell. Mol. Life Sci. CMLS 2000, 57, 235–249. [Google Scholar] [CrossRef]
- Nissim-Rafinia, M.; Kerem, B. Splicing regulation as a potential genetic modifier. Trends Genet. TIG 2002, 18, 123–127. [Google Scholar] [CrossRef]
- Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar] [CrossRef]
- Ma, L.; Li, A.; Zou, D.; Xu, X.; Xia, L.; Yu, J.; Bajic, V.B.; Zhang, Z. LncRNAWiki: Harnessing community knowledge in collaborative curation of human long non-coding RNAs. Nucleic Acids Res. 2015, 43, D187–D192. [Google Scholar] [CrossRef]
- Cao, M.; Zhao, J.; Hu, G. Genome-wide methods for investigating long noncoding RNAs. Biomed. Pharm. 2018, 111, 395–401. [Google Scholar] [CrossRef]
- Khalil, A.M.; Guttman, M.; Huarte, M.; Garber, M.; Raj, A.; Rivea Morales, D.; Thomas, K.; Presser, A.; Bernstein, B.E.; van Oudenaarden, A.; et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc. Natl. Acad. Sci. USA 2009, 106, 11667–11672. [Google Scholar] [CrossRef]
- Werner, M.S.; Sullivan, M.A.; Shah, R.N.; Nadadur, R.D.; Grzybowski, A.T.; Galat, V.; Moskowitz, I.P.; Ruthenburg, A.J. Chromatin-enriched lncRNAs can act as cell-type specific activators of proximal gene transcription. Nat. Struct. Mol. Biol. 2017, 24, 596–603. [Google Scholar] [CrossRef]
- Juarez-Mendez, S.; Zentella-Dehesa, A.; Villegas-Ruiz, V.; Perez-Gonzalez, O.A.; Salcedo, M.; Lopez-Romero, R.; Roman-Basaure, E.; Lazos-Ochoa, M.; Montes de Oca-Fuentes, V.E.; Vazquez-Ortiz, G.; et al. Splice variants of zinc finger protein 695 mRNA associated to ovarian cancer. J. Ovarian Res. 2013, 6, 61. [Google Scholar] [CrossRef]
- Nilsen, T.W.; Graveley, B.R. Expansion of the eukaryotic proteome by alternative splicing. Nature 2010, 463, 457–463. [Google Scholar] [CrossRef]
- Venables, J.P. Aberrant and alternative splicing in cancer. Cancer Res. 2004, 64, 7647–7654. [Google Scholar] [CrossRef]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M. Treatment of estrogen receptor-positive breast cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef]
- Harrow, J.; Frankish, A.; Gonzalez, J.M.; Tapanari, E.; Diekhans, M.; Kokocinski, F.; Aken, B.L.; Barrell, D.; Zadissa, A.; Searle, S.; et al. GENCODE: The reference human genome annotation for The ENCODE Project. Genome Res. 2012, 22, 1760–1774. [Google Scholar] [CrossRef]
- Nishikawa, T.; Ota, T.; Kawai, Y.; Ishii, S.; Saito, K.; Yamamoto, J.; Wakamatsu, A.; Ozawa, M.; Suzuki, Y.; Sugano, S.; et al. Database and analysis system for cDNA clones obtained from full-length enriched cDNA libraries. Silico Biol. 2002, 2, 5–18. [Google Scholar]
- Vorlova, S.; Rocco, G.; Lefave, C.V.; Jodelka, F.M.; Hess, K.; Hastings, M.L.; Henke, E.; Cartegni, L. Induction of antagonistic soluble decoy receptor tyrosine kinases by intronic polyA activation. Mol. Cell 2011, 43, 927–939. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Huveldt, D.; Kreinest, P.; Lohse, C.M.; Cheville, J.C.; Parker, A.S.; Copland, J.A.; Anastasiadis, P.Z. A p120 catenin isoform switch affects Rho activity, induces tumor cell invasion, and predicts metastatic disease. J. Biol. Chem. 2008, 283, 18344–18354. [Google Scholar] [CrossRef]
- Karni, R.; de Stanchina, E.; Lowe, S.W.; Sinha, R.; Mu, D.; Krainer, A.R. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat. Struct. Mol. Biol. 2007, 14, 185–193. [Google Scholar] [CrossRef]
- Jung, H.; Lee, D.; Lee, J.; Park, D.; Kim, Y.J.; Park, W.Y.; Hong, D.; Park, P.J.; Lee, E. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat. Genet. 2015, 47, 1242–1248. [Google Scholar] [CrossRef]
- Li, R.; Campos, J.; Iida, J. A gene regulatory program in human breast cancer. Genetics 2015, 201, 1341–1348. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamahsita, S.; Matsuda, Y.; Kishino, T.; Nakajima, T.; Kushima, R.; Kato, K.; Igaki, H.; Tachimori, Y.; Osugi, H.; et al. ZNF695 methylation predicts a response of esophageal squamous cell carcinoma to definitive chemoradiotherapy. J. Cancer Res. Clin. Oncol. 2015, 141, 453–463. [Google Scholar] [CrossRef]
- Li, C.; Kuang, L.; Zhu, B.; Chen, J.; Wang, X.; Huang, X. Identification of prognostic risk factors of acute lymphoblastic leukemia based on mRNA expression profiling. Neoplasma 2017, 64, 494–501. [Google Scholar] [CrossRef]
- Grondin, B.; Bazinet, M.; Aubry, M. The KRAB zinc finger gene ZNF74 encodes an RNA-binding protein tightly associated with the nuclear matrix. J. Biol. Chem. 1996, 271, 15458–15467. [Google Scholar] [CrossRef]
- Stein, L.D. Human genome: End of the beginning. Nature 2004, 431, 915–916. [Google Scholar] [CrossRef]
- Ponting, C.P.; Belgard, T.G. Transcribed dark matter: Meaning or myth? Hum. Mol. Genet. 2010, 19, R162–R168. [Google Scholar] [CrossRef]
- Brosnan, C.A.; Voinnet, O. The long and the short of noncoding RNAs. Curr. Opin. Cell Biol. 2009, 21, 416–425. [Google Scholar] [CrossRef]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef]
- Ono, H.; Motoi, N.; Nagano, H.; Miyauchi, E.; Ushijima, M.; Matsuura, M.; Okumura, S.; Nishio, M.; Hirose, T.; Inase, N.; et al. Long noncoding RNA HOTAIR is relevant to cellular proliferation, invasiveness, and clinical relapse in small-cell lung cancer. Cancer Med. 2014, 3, 632–642. [Google Scholar] [CrossRef]
- Guo, F.; Guo, L.; Li, Y.; Zhou, Q.; Li, Z. MALAT1 is an oncogenic long non-coding RNA associated with tumor invasion in non-small cell lung cancer regulated by DNA methylation. Int. J. Clin. Exp. Pathol. 2015, 8, 15903–15910. [Google Scholar]
- Li, J.; Chen, Y.; Chen, Z.; He, A.; Xie, H.; Zhang, Q.; Cai, Z.; Liu, Y.; Huang, W. SPRY4-IT1: A novel oncogenic long non-coding RNA in human cancers. Tumour Biol. 2017, 39, 1010428317711406. [Google Scholar] [CrossRef]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 14, 184. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcript | Primer Sequence | TM | Amplicon Size |
|---|---|---|---|
| RPL4 | Forward: 5′ CGAATGAGAGCTGGCAAAGGCAAA 3′ | ||
| Reverse: 5′ ACGCCAAGTGCCGTACAATTCATC 3′ | 60 °C | 243 bp | |
| ZNF695_TV1 | Forward: 5′ CTGGAGAGGCTCTTTGTACTTG 3′ | ||
| Reverse: 5′ GATAGGTTAACGTTGGTGGTAGG 3′ | 57.2 °C | 279 bp | |
| ZNF695 TV2/TV3 | Forward: 5′CCTTTGCCTTCTGCCATGAT 3′ | ||
| Reverse: 5′ TTAATTCAGAACTCGGGCTGAC 3′ | 55.2 °C | 212 bp | |
| ZNF695_TV4 | Forward: 5′ GTGGCCTGCAGGGACTATTG 3′ | ||
| Reverse: 5′ TGCAAGAGACATTGCCACATTC 3′ | 56.5 °C | 591 bp | |
| ZNF695_TV5 | Forward: 5′ CTCCCTTGGTCTTGCTATGT 3′ | ||
| Reverse: 5′ TGCAAGAGACATTGCCACATTC 3′ | 54.5 °C | 425 bp | |
| ZNF695_TV6 | Forward: 5′ GTGGCCTGCAGGGACTATTG 3′ | ||
| Reverse: 5′ TTAATTCAGAACTCGGGCTGAC 3′ | 56.5 °C | 638 bp | |
| ZNF695_TV7 | Forward: 5′ CTCCCTTGGTCTTGCTATGT 3′ | ||
| Reverse: 5′ TTAATTCAGAACTCGGGCTGAC 3′ | 54.5 °C | 472 bp |
| ZNF695 Splice Variant Expression | |||||||
|---|---|---|---|---|---|---|---|
| Age | Total Patients | ZNF695_TV1 | ZNF695_TV3 | ZNF695_TV4 | ZNF695_TV5 | ZNF695_TV6 | ZNF695_TV7 |
| <10 years | 14 (32.5%) | 6 (40%) | 4 (37.7%) | 4 (37.7%) | 3 (20%) | 2 (13.4%) | 2 (13.4%) |
| >10 years | 29 (67.5%) | 16 (57.2%) | 14 (50%) | 12 (42.9%) | 4 (14.3%) | 1 (3.6%) | 4 (14.3%) |
| Sex | |||||||
| Male | 22 (51.1%) | 8 (40%) | 7 (35%) | 5 (25%) | 1 (5%) | 1 (5%) | 2 (10%) |
| Female | 21 (48.9%) | 14 (60.9%) | 11 (38%) | 11 (38%) | 6 (20.7%) | 2 (6.9%) | 4 (13.8%) |
| Leukocytes | |||||||
| <50,000 | 32(74.4%) | 17 (53.2%) | 15 (46.8%) | 11 (34.4%) | 4 (15.7%) | 3 (9.4%) | 4 (15.7%) |
| >50,000 | 11 (25.6%) | 5 (45. 6%) | 3 (27.3%) | 5 (45. 6%) | 2 (18.2%) | 0 (0%) | 1 (9.1%) |
| Hemoglobin | |||||||
| <10 g/dL | 33 (76.7%) | 17 (51.6%) | 14 (42.5%) | 13 (39.4%) | 7 (21.3%) | 3 (9.1%) | 5 (18.2%) |
| > or = 10 g/dL | 10 (23.3%) | 5 (50%) | 4 (40%) | 3 (30%) | 0 (0%) | 0 (0%) | 1 (10%) |
| Hypodiploidy | |||||||
| Positive | 10 (23.2%) | 4 (40%) | 4 (40%) | 5 (50%) | 2 (20%) | 0 (0%) | 1 (10%) |
| Negative | 33 (76.8%) | 18 (54.6%) | 14 (42.5%) | 12 (36.4%) | 5 (15.2%) | 3 (9.1%) | 5 (15.2%) |
| Philadelphia chromosome | |||||||
| Positive | 2 (4.6%) | 2 (100%) | 2 (100%) | 2 (100%) | 1 (50%) | 0 (0%) | 1 (50%) |
| Negative | 41 (95.4%) | 20 (48.8%) | 16 (39.1%) | 14 (34.2%) | 6 (14.6%) | 3 (7.4%) | 5 (12.2%) |
| Relapse | |||||||
| Positive | 15 (34.8%) | 8 (53.4%) | 8 (53.4%) | 7 (46.6%) | 2 (13.4%) | 1 (6.7%) | 3 (20%) |
| Negative | 27 (65.2%) | 14 (51.9%) | 11 (40.8%) | 9 (33.4%) | 5 (18.6%) | 2 (7.5%) | 3 (11.2%) |
| Survival | |||||||
| Positive | 34 (79%) | 15 (44.9%) | 12 (35.3%) | 11 (32.4%) | 6 (17.7%) | 3 (8.9%) | 5 (14.7%) |
| Negative | 9 (21%) | 7 (77.8%) | 6(66.7%) | 5 (44.5%) | 1 (11.2%) | 0 (0%) | 1 (11.2%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De la Rosa, R.; Villegas-Ruíz, V.; Caballero-Palacios, M.C.; Pérez-López, E.I.; Murata, C.; Zapata-Tarres, M.; Cárdenas-Cardos, R.; Paredes-Aguilera, R.; Rivera-Luna, R.; Juárez-Méndez, S. Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia. Genes 2019, 10, 716. https://doi.org/10.3390/genes10090716
De la Rosa R, Villegas-Ruíz V, Caballero-Palacios MC, Pérez-López EI, Murata C, Zapata-Tarres M, Cárdenas-Cardos R, Paredes-Aguilera R, Rivera-Luna R, Juárez-Méndez S. Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia. Genes. 2019; 10(9):716. https://doi.org/10.3390/genes10090716
Chicago/Turabian StyleDe la Rosa, Ricardo, Vanessa Villegas-Ruíz, Marcela Concepción Caballero-Palacios, Eleazar Israel Pérez-López, Chiharu Murata, Martha Zapata-Tarres, Rocio Cárdenas-Cardos, Rogelio Paredes-Aguilera, Roberto Rivera-Luna, and Sergio Juárez-Méndez. 2019. "Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia" Genes 10, no. 9: 716. https://doi.org/10.3390/genes10090716
APA StyleDe la Rosa, R., Villegas-Ruíz, V., Caballero-Palacios, M. C., Pérez-López, E. I., Murata, C., Zapata-Tarres, M., Cárdenas-Cardos, R., Paredes-Aguilera, R., Rivera-Luna, R., & Juárez-Méndez, S. (2019). Expression of ZNF695 Transcript Variants in Childhood B-Cell Acute Lymphoblastic Leukemia. Genes, 10(9), 716. https://doi.org/10.3390/genes10090716