Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations

Abstract
1. Introduction
2. Patients and Methods
2.1. Subjects
2.2. RET Genetic Analysis
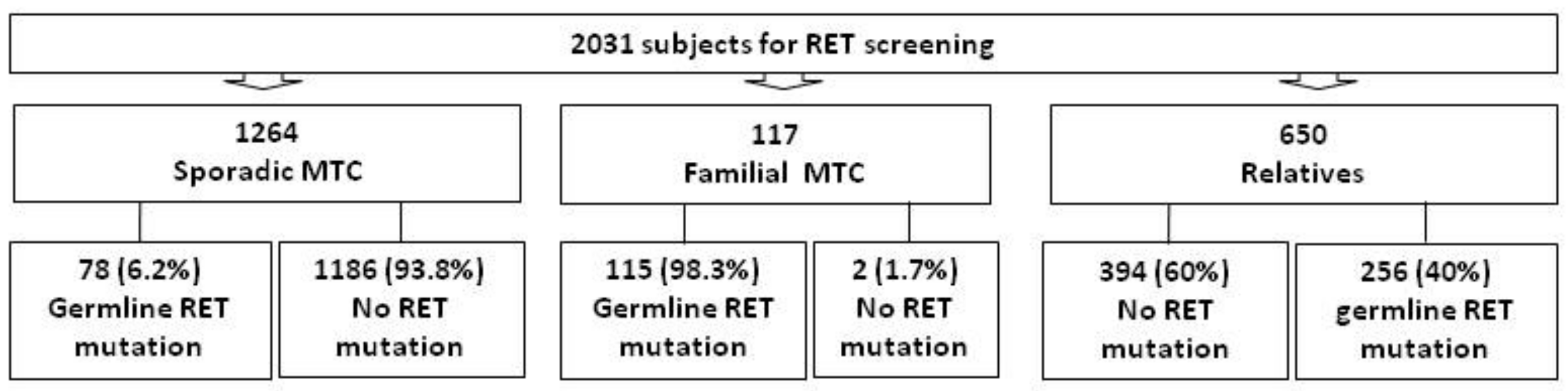
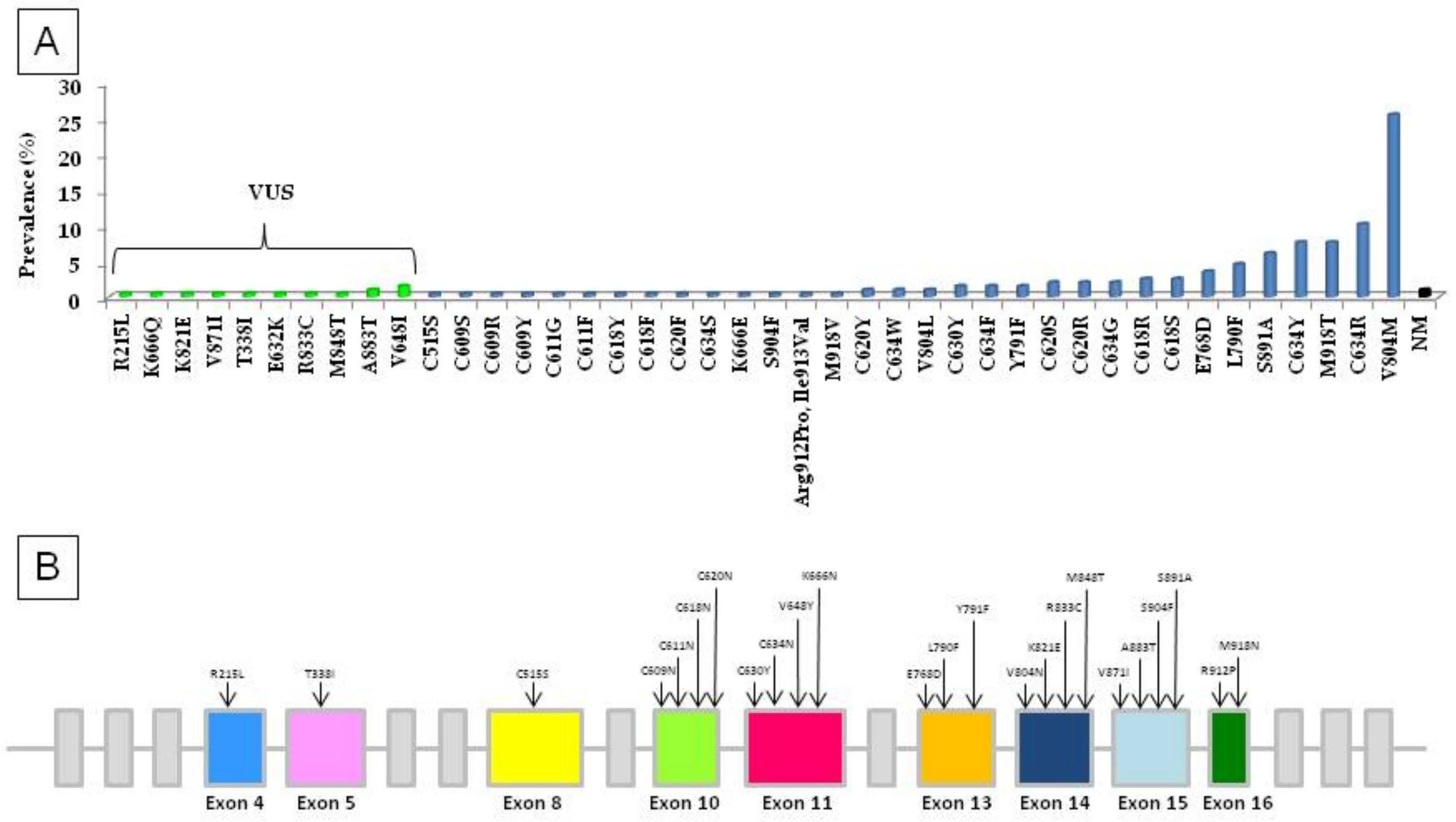
3. Results
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Romei, C.; Pardi, E.; Cetani, F.; Elisei, R. Genetic and clinical features of multiple endocrine neoplasia types 1 and 2. J. Oncol. 2012, 2012, 15. [Google Scholar] [CrossRef]
- Wohllk, N.; Schweizer, H.; Erlic, Z.; Schmid, K.W.; Walz, M.K.; Raue, F.; Neumann, H.P.H. Multiple endocrine neoplasia type 2. Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Front. Physiol. 2018, 9, 1873. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M.; Kwok, J.B.J.; Healey, C.S.; Elsdon, M.J.; Eng, C.; Gardner, E.; Love, D.R.; Mole, S.E.; Moore, J.K.; Papi, L.; et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.A.J. Advances in the management of MEN2: From improved surgical and medical treatment to novel kinase inhibitors. Endocr. Relat. Cancer 2018, 25, T1–T13. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef]
- Hofstra, R.M.W.; Landsvater, R.M.; Ceccherini, I.; Stulp, R.P.; Stelwagen, T.; Luo, Y.; Pasini, B.; Höppener, J.W.M.; Van Amstel, H.K.P.; Romeo, G.; et al. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994, 367, 375–376. [Google Scholar] [CrossRef]
- Antinolo, G.; Marcos, I.; Fernandez, R.M.; Romero, M.; Borrego, S. A novel germline point mutation, c.2304 G-->T, in codon 768 of the RET proto-oncogene in a patient with medullary thyroid carcinoma. Am. J. Med. Genet. 2002, 110, 85–87. [Google Scholar] [CrossRef]
- Berndt, I.; Reuter, M.; Saller, B.; Frank-Raue, K.; Groth, P.; Grussendorf, M.; Raue, F.; Ritter, M.M.; Hoppner, W. A New Hot Spot for Mutations in the ret Protooncogene Causing Familial Medullary Thyroid Carcinoma and Multiple Endocrine Neoplasia Type 2A. J. Clin. Endocrinol. Metab. 1998, 83, 770–774. [Google Scholar] [CrossRef]
- Fink, M.; Weinhäusel, A.; Niederle, B.; Haas, O.A. Distinction between sporadic and hereditary medullary thyroid carcinoma (MTC) by mutation analysis of the RET proto-oncogene. Int. J. Cancer 1996, 69, 312–316. [Google Scholar] [CrossRef]
- Marsh, D.J.; Learoyd, D.L.; Andrew, S.D.; Krishnan, L.; Pojer, R.; Richardson, A.L.; Delbridge, L.; Eng, C.; Robinson, B.G. Somatic mutations in the RET proto-oncogene in sporadic medullary thyroid carcinoma. Clin. Endocrinol. 1996, 44, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Dang, G.T.; Cote, G.J.; Schultz, P.N.; Khorana, S.; Decker, R.A.; Gagel, R.F. A codon 891 exon 15 RET proto-oncogene mutation in familial medullary thyroid carcinoma: A detection strategy. Mol. Cell. Probes 1999, 13, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Crockett, D.K.; Piccolo, S.R.; Ridge, P.G.; Margraf, R.L.; Lyon, E.; Williams, M.S.; Mitchell, J.A. Predicting Phenotypic Severity of Uncertain Gene Variants in the RET Proto-Oncogene. PLoS ONE 2011, 6, e18380. [Google Scholar] [CrossRef] [PubMed]
- Cosci, B.; Vivaldi, A.; Romei, C.; Gemignani, F.; Landi, S.; Ciampi, R.; Tacito, A.; Molinaro, E.; Agate, L.; Bottici, V.; et al. In silico and in vitro analysis of rare germline allelic variants of RET oncogene associated with medullary thyroid cancer. Endocr. Relat. Cancer 2011, 18, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Margraf, R.L.; Crockett, D.K.; Krautscheid, P.M.F.; Seamons, R.; Calderon, F.R.O.; Wittwer, C.T.; Mao, R. Multiple endocrine neoplasia type 2 RET protooncogene database: Repository of MEN2-associated RET sequence variation and reference for genotype/phenotype correlations. Hum. Mutat. 2009, 30, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Raue, F.; Frank-Raue, K. Update on Multiple Endocrine Neoplasia Type 2: Focus on Medullary Thyroid Carcinoma. J. Endocr. Soc. 2018, 2, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Cosci, B.; Renzini, G.; Bottici, V.; Agate, L.; Passannanti, P.; Viola, D.; Biagini, A.; Materazzi, G.; Pinchera, A.; et al. RET genetic screening of sporadic medullary thyroid cancer (MTC) allows the preclinical diagnosis of unsuspected gene carriers and the identification of a relevant percentage of hidden familial MTC (FMTC). Clin. Endocrinol. 2011, 74, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Wiench, M.; Wygoda, Z.; Gubala, E.; Wloch, J.; Lisowska, K.; Krassowski, J.; Scieglinska, D.; Fiszer-Kierzkowska, A.; Lange, D.; Kula, D.; et al. Estimation of Risk of Inherited Medullary Thyroid Carcinoma in Apparent Sporadic Patients. J. Clin. Oncol. 2001, 19, 1374–1380. [Google Scholar] [CrossRef]
- Niccoli-Sire, P.; Murat, A.; Rohmer, V.; Franc, S.; Chabrier, G.; Baldet, L.; Maes, B.; Savagner, F.; Giraud, S.; Bezieau, S.; et al. Familial Medullary Thyroid Carcinoma with Noncysteine RET Mutations: Phenotype-Genotype Relationship in a Large Series of Patients. J. Clin. Endocrinol. Metab. 2001, 86, 3746–3753. [Google Scholar] [CrossRef]
- Catalogue of Somatic Mutations in Cancer. Available online: https://cancer.sanger.ac.uk (accessed on 13 June 2019).
- Romei, C.; Ugolini, C.; Cosci, B.; Torregrossa, L.; Vivaldi, A.; Ciampi, R.; Tacito, A.; Basolo, F.; Materazzi, G.; Miccoli, P.; et al. Low Prevalence of the Somatic M918T RET Mutation in Micro-Medullary Thyroid Cancer. Thyroid 2012, 22, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Romei, C.; Casella, F.; Tacito, A.; Bottici, V.; Valerio, L.; Viola, D.; Cappagli, V.; Matrone, A.; Ciampi, R.; Piaggi, P.; et al. New insights in the molecular signature of advanced medullary thyroid cancer: Evidence of a bad outcome of cases with double RET mutations. J. Med. Genet. 2016, 53, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.M.B.; Camacho, C.P.; Assumpção, L.V.M.; Bufalo, N.E.; Carvalho, A.L.; De Carvalho, G.A.; Castroneves, L.A.; De Castro, F.M.; Ceolin, L.; Cerutti, J.M.; et al. Genotype and phenotype landscape of MEN2 in 554 medullary thyroid cancer patients: The BrasMEN study. Endocr. Connect. 2019, 8, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Machens, A.; Hoegel, J.; Van Vroonhoven, T.J.; Roeher, H.-D.; Wahl, R.A.; Raue, F.; Niccoli-Sire, P.; Frank-Raue, K.; Lamesch, P.; Conte-Devolx, B.; et al. Early Malignant Progression of Hereditary Medullary Thyroid Cancer. N. Engl. J. Med. 2003, 349, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Sarika, H.L.; Papathoma, A.; Garofalaki, M.; Vasileiou, V.; Vlassopoulou, B.; Anastasiou, E.; Alevizaki, M. High prevalence of exon 8 G533C mutation in apparently sporadic medullary thyroid carcinoma in Greece. Clin. Endocrinol. 2012, 77, 857–862. [Google Scholar] [CrossRef]
- Fanis, P.; Skordis, N.; Frangos, S.; Christopoulos, G.; Spanou-Aristidou, E.; Andreou, E.; Manoli, P.; Mavrommatis, M.; Nicolaou, S.; Kleanthous, M.; et al. Multiple endocrine neoplasia 2 in Cyprus: Evidence for a founder effect. J. Endocrinol. Investig. 2018, 41, 1149–1157. [Google Scholar] [CrossRef]
- Romei, C.; Mariotti, S.; Fugazzola, L.; Taccaliti, A.; Pacini, F.; Opocher, G.; Mian, C.; Castellano, M.; Degli Uberti, E.; Ceccherini, I.; et al. Multiple endocrine neoplasia type 2 syndromes (MEN 2): Results from the ItaMEN network analysis on the prevalence of different genotypes and phenotypes. Eur. J. Endocrinol. 2010, 163, 301–308. [Google Scholar] [CrossRef]
- Wells, S.A.J.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef]
- Elisei, R.; Alevizaki, M.; Conte-Devolx, B.; Frank-Raue, K.; Leite, V.; Williams, G. 2012 European Thyroid Association Guidelines for Genetic Testing and Its Clinical Consequences in Medullary Thyroid Cancer. Eur. Thyroid J. 2012, 1, 216–231. [Google Scholar] [CrossRef]
- Elisei, R.; Romei, C.; Renzini, G.; Bottici, V.; Cosci, B.; Molinaro, E.; Agate, L.; Cappagli, V.; Miccoli, P.; Berti, P.; et al. The Timing of Total Thyroidectomy in RET Gene Mutation Carriers Could Be Personalized and Safely Planned on the Basis of Serum Calcitonin: 18 Years Experience at One Single Center. J. Clin. Endocrinol. Metab. 2012, 97, 426–435. [Google Scholar] [CrossRef]
- Elisei, R.; Cosci, B.; Romei, C.; Bottici, V.; Sculli, M.; Lari, R.; Barale, R.; Pacini, F.; Pinchera, A. RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J. Clin. Endocrinol. Metab. 2004, 89, 3579–3584. [Google Scholar] [CrossRef] [PubMed]
- Pecce, V.; Sponziello, M.; Damante, G.; Rosignolo, F.; Durante, C.; Lamartina, L.; Grani, G.; Russo, D.; di Gioia, C.R.; Filetti, S.; et al. A synonymous RET substitution enhances the oncogenic effect of an in-cis missense mutation by increasing constitutive splicing efficiency. PLoS Genet. 2018, 14, e1007678. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.; Mulligan, L.M.; Smith, D.P.; Healey, C.S.; Frilling, A.; Raue, F.; Neumann, H.P.; Ponder, M.A.; Ponder, B.A. Low frequency of germline mutations in the RET proto-oncogene in patients with apparently sporadic medullary thyroid carcinoma. Clin. Endocrinol. 1995, 43, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Eng, C. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996, 276, 1575–1579. [Google Scholar] [CrossRef] [PubMed]
- Pinna, G.; Orgiana, G.; Riola, A.; Ghiani, M.; Lai, M.L.; Carcassi, C.; Mariotti, S. RET Proto-Oncogene in Sardinia: V804M Is the Most Frequent Mutation and May Be Associated with FMTC/MEN-2A Phenotype. Thyroid 2007, 17, 101–104. [Google Scholar] [CrossRef]
- Machens, A.; Dralle, H. Familial prevalence and age of RET germline mutations: Implications for screening. Clin. Endocrinol. 2008, 69, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Frank-Raue, K.; Raue, F. Hereditary Medullary Thyroid Cancer Genotype–Phenotype Correlation. Methods Mol. Biol. 2015, 204, 139–156. [Google Scholar]
- Toledo, R.A.; Maciel, R.M.B.; Erlic, Z.; Lourenco, D.M.J.; Cerutti, J.M.; Eng, C.; Neumann, H.P.; Toledo, S.P. RET Y791F Variant Does Not Increase the Risk for Medullary Thyroid Carcinoma. Thyroid 2015, 25, 973–974. [Google Scholar] [CrossRef]
- Nagal, M.A.; Healey, C.S.; Ponder, M.A.; Gardner, E.; Scheumann, G.F.; Jackson, C.E.; Tunnacllffe, A.; Eng, C.; Smith, D.P.; Mulligan, L.M. Point mutation within the tyrosine kinase domain of the RET proto-oncogene in multiple endocrine neoplasia type 2B and related sporadic tumours. Hum. Mol. Genet. 1994, 3, 237–241. [Google Scholar]
- Brauckhoff, M.; Machens, A.; Lorenz, K.; Bjoro, T.; Varhaug, J.E.; Dralle, H. Surgical curability of medullary thyroid cancer in multiple endocrine neoplasia 2B: A changing perspective. Ann. Surg. 2014, 259, 800–806. [Google Scholar] [CrossRef]
- Carlson, K.M.; Bracamontes, J.; Jackson, C.E.; Clark, R.; Lacroix, A.; Wells, S.A.; Goodfellow, P.J. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am. J. Hum. Genet. 1994, 55, 1076–1082. [Google Scholar] [PubMed]
- Raue, F.; Frank-Raue, K. Genotype-phenotype correlation in multiple endocrine neoplasia type 2. Clinics (Sao Paulo) 2012, 67, 69–75. [Google Scholar] [CrossRef]
- Santoro, M.; Carlomagno, F.; Romano, A.; Bottaro, D.; Dathan, N.; Grieco, M.; Fusco, A.; Vecchio, G.; Matoskova, B.; Kraus, M.; et al. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science 1995, 267, 381–383. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, G.; Ceccherini, I.; Lerone, M.; Jasonni, V. Pathogenesis of Hirschsprung’s disease. J. Pediatr. Surg. 2000, 35, 1017–1025. [Google Scholar] [CrossRef] [PubMed]
- Amiel, J.; Sproat-Emison, E.; Garcia-Barcelo, M.; Lantieri, F.; Burzynski, G.; Borrego, S.; Pelet, A.; Arnold, S.; Miao, X.; Griseri, P.; et al. Hirschsprung disease, associated syndromes and genetics: A review. J. Med. Genet. 2008, 45, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Arighi, E.; Popsueva, A.; Degl’Innocenti, D.; Borrello, M.G.; Carniti, C.; Perälä, N.M.; Pierotti, M.A.; Sariola, H. Biological Effects of the Dual Phenotypic Janus Mutation of ret Cosegregating with Both Multiple Endocrine Neoplasia Type 2 and Hirschsprung’s Disease. Mol. Endocrinol. 2004, 18, 1004–1017. [Google Scholar] [CrossRef] [PubMed][Green Version]




{kind=link}
{kind=link}
{kind=link}
| RET Mutation | Families | PATIENTS (n.) | In Silico Prediction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Studied | RET Positive | RET Negative | Affected | Gene Carriers | SIFT | POLYPHEN | CADD | REVEL | META LR | MUTATION ASSESSOR | ||
| R215L a | 1 | 1 | 1 | 0 | 1 | 0 | tolerated | likely benign | likely benign | benign | tolerated | 0.medium |
| K666Q | 1 | 1 | 1 | 0 | 1 | 0 | deleterious | possibly damaging | likely benign | Likely disease causing | damaging | low |
| K821E | 1 | 3 | 3 | 0 | 1 | 2 | deleterious | probably damaging | likely benign | Likely disease causing | damaging | low |
| V871I | 1 | 4 | 3 | 1 | 1 | 2 | deleterious | probably damaging | likely benign | Likely disease causing | damaging | neutral |
| T338I | 1 | 3 | 2 | 1 | 1 | 1 | tolerated | benign | likely benign | likely benign | tolerated | medium |
| E632K | 1 | 1 | 1 | 0 | 1 | 0 | tolerated | possibly damaging | likely benign | Likely disease causing | damaging | medium |
| V648I | 2 | 6 | 2 | 4 | 1 | 1 | tolerated | benign | likely benign | Likely disease causing | damaging | neutral |
| R833C | 1 | 2 | 2 | 0 | 1 | 1 | deleterious | probably damaging | likely benign | Likely disease causing | damaging | medium |
| M848T | 1 | 2 | 2 | 0 | 1 | 1 | tolerated | probably damaging | likely benign | Likely disease causing | damaging | neutral |
| A883T | 2 | 14 | 9 | 5 | 3 | 7 | deleterious | probably damaging | likely deleterious | Likely disease causing | tolerated | neutral |
| RET Mutation | Number of Families | Number of Families with PHEO/Number of Total Families | Number of Families with hyperPTH/Number of Total Families | Number of Families with Other Diseases/NUMBER of Total Families |
|---|---|---|---|---|
| V648I | 3 | 0/3 | 0/3 | 1/3 a |
| E768D | 7 | 0/7 | 0/7 | 1/7 b |
| V804M | 50 | 2/50 | 1/50 | 12/50 a |
| S891A | 12 | 0/12 | 0/12 | 2/12 b,1/12 d |
| C609R | 1 | 0/1 | 0/1 | 1/1 b |
| C6111F | 1 | 0/1 | 1/1 | 0/1 |
| C618R | 5 | 2/5 | 0/5 | 1/5 b |
| C618S | 5 | 1/5 | 0/5 | 0/5 |
| C620R | 4 | 1/4 | 0/4 | 1/4 a |
| C620S | 4 | 0/4 | 0/4 | 1/4 a |
| C620Y | 2 | 0/2 | 0/2 | 1/2 b, 1/2 a |
| C630Y | 3 | 0/3 | 1/3 | 1/3 c, a |
| C634F | 3 | 3/3 | 0/3 | 1/3 c |
| C634G | 4 | 2/4 | 0/4 | 1/4 c |
| C634R | 20 | 13/20 | 3/20 | 2/20 c |
| C634S | 1 | 1/1 | 0/1 | 0/1 |
| C634Y | 15 | 11/15 | 3/15 | 1/15 c |
| C634W | 2 | 2/2 | 0/2 | 0/2 |
| M918T | 15 | 4/15 | 0/15 | 15/15 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elisei, R.; Tacito, A.; Ramone, T.; Ciampi, R.; Bottici, V.; Cappagli, V.; Viola, D.; Matrone, A.; Lorusso, L.; Valerio, L.; et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes 2019, 10, 698. https://doi.org/10.3390/genes10090698
Elisei R, Tacito A, Ramone T, Ciampi R, Bottici V, Cappagli V, Viola D, Matrone A, Lorusso L, Valerio L, et al. Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes. 2019; 10(9):698. https://doi.org/10.3390/genes10090698
Chicago/Turabian StyleElisei, Rossella, Alessia Tacito, Teresa Ramone, Raffaele Ciampi, Valeria Bottici, Virginia Cappagli, David Viola, Antonio Matrone, Loredana Lorusso, Laura Valerio, and et al. 2019. "Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations" Genes 10, no. 9: 698. https://doi.org/10.3390/genes10090698
APA StyleElisei, R., Tacito, A., Ramone, T., Ciampi, R., Bottici, V., Cappagli, V., Viola, D., Matrone, A., Lorusso, L., Valerio, L., Giani, C., Campopiano, C., Prete, A., Agate, L., Molinaro, E., & Romei, C. (2019). Twenty-Five Years Experience on RET Genetic Screening on Hereditary MTC: An Update on The Prevalence of Germline RET Mutations. Genes, 10(9), 698. https://doi.org/10.3390/genes10090698