Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges




Abstract
1. Introduction
1.1. Inherited Retinal Diseases (IRDs)
1.2. Present Treatment of IRD
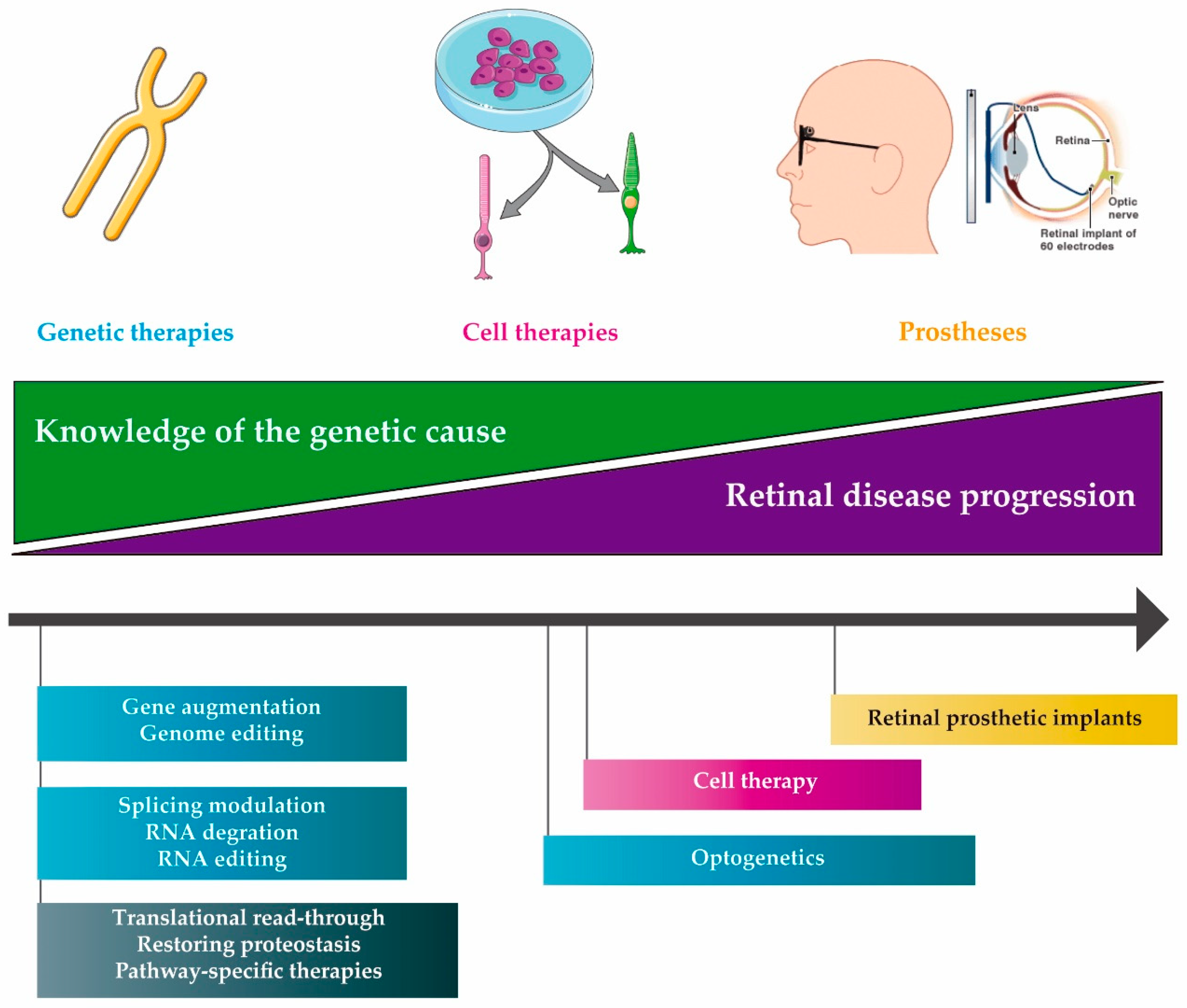
2. Molecular therapies
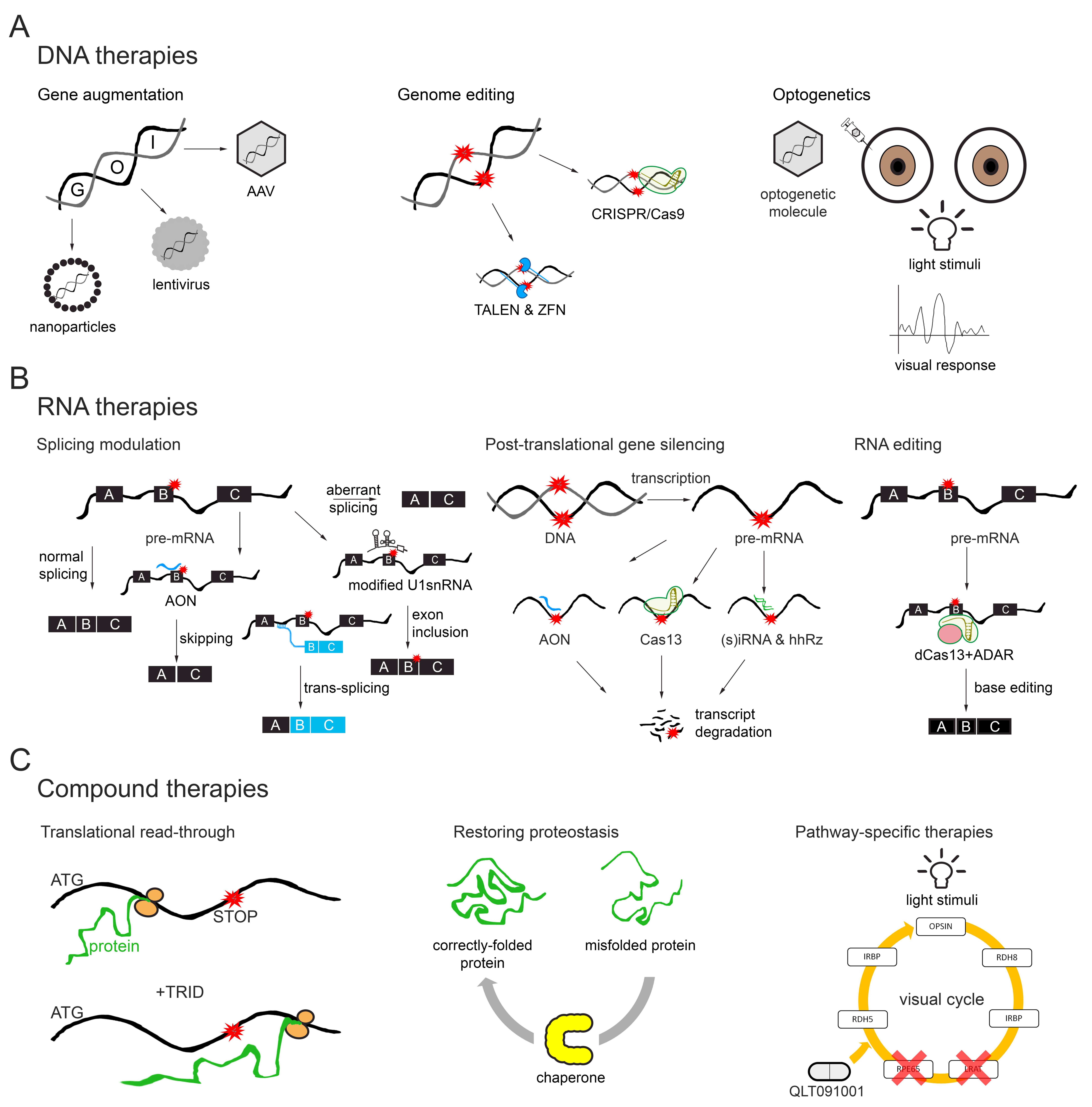
2.1. DNA Therapies
2.1.1. Gene Augmentation
2.1.2. Genome Editing
2.1.3. Optogenetics
2.2. RNA Therapies
2.2.1. Splicing Modulation
2.2.2. Post-transcriptional Gene Silencing
2.2.3. RNA Editing (dCas13 and ADAR)
2.3. Compound Therapies
2.3.1. Translational Read-Through
2.3.2. Restoring Proteostasis (Protein Therapies)
2.3.3. Pathway-Specific Therapies
3. Delivery of Therapeutic Molecules
3.1. Methods of Ocular Delivery
3.2. Vectors
3.2.1. Viral Vectors
3.2.2. Non-Viral Vectors
4. Other IRD Treatments
4.1. Cell Therapy
4.2. Retinal Prosthetic Implants
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Saperstein, D.A. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef]
- Khan, M.; Fadaie, Z.; Cornelis, S.S.; Cremers, F.P.; Roosing, S. Identification and Analysis of Genes Associated with Inherited Retinal Diseases. Methods Mol. Biol. 2019, 1834, 3–27. [Google Scholar] [PubMed]
- Kaur, C.; Foulds, W.; Ling, E. Blood–retinal barrier in hypoxic ischaemic conditions: Basic concepts, clinical features and management. Prog. Retin. Eye Res. 2008, 27, 622–647. [Google Scholar] [CrossRef] [PubMed]
- Anand, V.; Duffy, B.; Yang, Z.; Dejneka, N.S.; Maguire, A.M.; Bennett, J. A Deviant Immune Response to Viral Proteins and Transgene Product Is Generated on Subretinal Administration of Adenovirus and Adeno-associated Virus. Mol. Ther. 2002, 5, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J. Immune response following intraocular delivery of recombinant viral vectors. Gene Ther. 2003, 10, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Öner, A. Recent Advancements in Gene Therapy for Hereditary Retinal Dystrophies. Turk. J. Ophthalmol. 2017, 47, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.R.; Huckfeldt, R.M. Gene therapy for inherited retinal degenerations: Initial successes and future challenges. J. Neural Eng. 2017, 14, 051002. [Google Scholar] [CrossRef]
- Liang, F.Q.; Anand, V.; Maguire, A.M.; Bennett, J. Intraocular delivery of recombinant virus. Methods Mol. Med. 2001, 47, 125–139. [Google Scholar]
- Ong, T.; Pennesi, M.E.; Birch, D.G.; Lam, B.L.; Tsang, S.H. Adeno-Associated Viral Gene Therapy for Inherited Retinal Disease. Pharm. Res. 2019, 36, 34. [Google Scholar] [CrossRef]
- Sahel, J.-A.; Marazova, K.; Audo, I. Clinical Characteristics and Current Therapies for Inherited Retinal Degenerations. Cold Spring Harb. Perspect. Med. 2014, 5, a017111. [Google Scholar] [CrossRef]
- Robson, A.G. Pattern ERG Correlates of Abnormal Fundus Autofluorescence in Patients with Retinitis Pigmentosa and Normal Visual Acuity. Investig. Opthalmology Vis. Sci. 2003, 44, 3544–3550. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Spaide, R.F.; Fisher, Y.L.; Freund, K.B.; Klancnik, J.M.; Yannuzzi, L.A. Three-Dimensional Evaluation of Vitreomacular Traction and Epiretinal Membrane Using Spectral-Domain Optical Coherence Tomography. Am. J. Ophthalmol. 2008, 145, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, A.; Yamamoto, S.; Ogata, K.; Sugawara, T.; Hiramatsu, A.; Shibata, M.; Mitamura, Y. Macular abnormalities in patients with retinitis pigmentosa: Prevalence on OCT examination and outcomes of vitreoretinal surgery. Acta Ophthalmol. 2011, 89, e122–e125. [Google Scholar] [CrossRef] [PubMed]
- Lewin, A.S.; Rossmiller, B.; Mao, H. Gene Augmentation for adRP Mutations in RHO. Cold Spring Harb. Perspect. Med. 2014, 4, a017400. [Google Scholar] [CrossRef] [PubMed]
- Salsman, J.; Dellaire, G. Precision genome editing in the CRISPR era. Biochem. Cell Boil. 2017, 95, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Mead, B.; Berry, M.; Logan, A.; Scott, R.A.; Leadbeater, W.; Scheven, B.A. Stem cell treatment of degenerative eye disease. Stem Cell Res. 2015, 14, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Weiland, J.D.; Humayun, M.S. Retinal prosthesis. IEEE Trans. Biomed. Eng. 2014, 61, 1412–1424. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Eye Res. 2010, 29, 335–375. [Google Scholar] [CrossRef]
- Smith, J.; Ward, D.; Michaelides, M.; Moore, A.T.; Simpson, S. New and emerging technologies for the treatment of inherited retinal diseases: A horizon scanning review. Eye 2015, 29, 1131–1140. [Google Scholar] [CrossRef]
- Schön, C.; Biel, M.; Michalakis, S. Retinal gene delivery by adeno-associated virus (AAV) vectors: Strategies and applications. Eur. J. Pharm. Biopharm. 2015, 95, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J. Taking Stock of Retinal Gene Therapy: Looking Back and Moving Forward. Mol. Ther. 2017, 25, 1076–1094. [Google Scholar] [CrossRef] [PubMed]
- Moore, N.A.; Morral, N.; Ciulla, T.A.; Bracha, P. Gene therapy for inherited retinal and optic nerve degenerations. Expert Opin. Biol. Ther. 2018, 18, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Zuris, J.A.; Thompson, D.B.; Shu, Y.; Guilinger, J.P.; Bessen, J.L.; Hu, J.H.; Liu, D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Arbabi, A.; Liu, A.; Ameri, H. Gene Therapy for Inherited Retinal Degeneration. J. Ocul. Pharmacol. Ther. 2019, 35, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Zangi, L.; Lui, K.O.; Von Gise, A.; Ma, Q.; Ebina, W.; Ptaszek, L.M.; Später, D.; Xu, H.; Tabebordbar, M.; Gorbatov, R.; et al. Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 2013, 31, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Ameri, H. Prospect of retinal gene therapy following commercialization of voretigene neparvovec-rzyl for retinal dystrophy mediated by RPE65 mutation. J. Curr. Ophthalmol. 2018, 30, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Rowe-Rendleman, C.L.; Durazo, S.A.; Kompella, U.B.; Rittenhouse, K.D.; Di Polo, A.; Weiner, A.L.; Grossniklaus, H.E.; Naash, M.I.; Lewin, A.S.; Horsager, A.; et al. Drug and Gene Delivery to the Back of the Eye: From Bench to Bedside. Investig. Opthalmol. Vis. Sci. 2014, 55, 2714–2730. [Google Scholar] [CrossRef] [PubMed]
- Soofiyani, S.R.; Baradaran, B.; Lotfipour, F.; Kazemi, T.; Mohammadnejad, L. Gene Therapy, Early Promises, Subsequent Problems, and Recent Breakthroughs. Adv. Pharm. Bull. 2013, 3, 249–255. [Google Scholar]
- Jacobson, S.G.; Cideciyan, A.V.; Roman, A.J.; Sumaroka, A.; Schwartz, S.B.; Héon, E.; Hauswirth, W.W. Improvement and Decline in Vision with Gene Therapy in Childhood Blindness. N. Engl. J. Med. 2015, 372, 1920–1926. [Google Scholar] [CrossRef]
- Gao, G.; Vandenberghe, L.H.; Wilson, J.M. New recombinant serotypes of AAV vectors. Curr. Gene Ther. 2005, 5, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Vandenberghe, L.H.; Wilson, J.M.; Gao, G. Tailoring the AAV vector capsid for gene therapy. Gene Ther. 2009, 16, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Le Meur, G.; Stieger, K.; Smith, A.J.; Weber, M.; Deschamps, J.Y.; Nivard, D.; Ali, R.R. Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007, 14, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.D.; McClements, M.E.; De La Camara, C.M.-F.; Bellingrath, J.-S.; Dauletbekov, D.; Ramsden, S.C.; Hickey, D.G.; Barnard, A.R.; MacLaren, R.E. Codon-Optimized RPGR Improves Stability and Efficacy of AAV8 Gene Therapy in Two Mouse Models of X-Linked Retinitis Pigmentosa. Mol. Ther. 2017, 25, 1854–1865. [Google Scholar] [CrossRef] [PubMed]
- Georgiadis, A.; Duran, Y.; Ribeiro, J.; Abelleira-Hervas, L.; Robbie, S.J.; Sünkel-Laing, B.; Fourali, S.; Cordero, A.G.; Cristante, E.; Michaelides, M.; et al. Development of an optimized AAV2/5 gene therapy vector for Leber congenital amaurosis owing to defects in RPE65. Gene Ther. 2016, 23, 857–862. [Google Scholar] [CrossRef] [PubMed]
- McClements, M.E.; MacLaren, R.E. Gene therapy for retinal disease. Transl. Res. 2013, 161, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.H.; Wensel, T.G. The Nature of Dominant Mutations of Rhodopsin and Implications for Gene Therapy. Mol. Neurobiol. 2003, 28, 149–158. [Google Scholar] [CrossRef]
- Farrar, G.J.; Millington-Ward, S.; Chadderton, N.; Humphries, P.; Kenna, P.F. Gene-based therapies for dominantly inherited retinopathies. Gene Ther. 2012, 19, 137–144. [Google Scholar] [CrossRef]
- O’Reilly, M.; Palfi, A.; Chadderton, N.; Millington-Ward, S.; Ader, M.; Cronin, T.; Tuohy, T.; Auricchio, A.; Hildinger, M.; Tivnan, A.; et al. RNA Interference–Mediated Suppression and Replacement of Human Rhodopsin In Vivo. Am. J. Hum. Genet. 2007, 81, 127–135. [Google Scholar] [CrossRef]
- Dyka, F.M.; Boye, S.L.; Chiodo, V.A.; Hauswirth, W.W.; Boye, S.E. Dual adeno-associated virus vectors result in efficient in vitro and in vivo expression of an oversized gene, MYO7A. Hum. Gene Ther. Methods 2014, 25, 166–177. [Google Scholar] [CrossRef]
- Trapani, I.; Colella, P.; Sommella, A.; Iodice, C.; Cesi, G.; de Simone, S.; Farrar, G.J. Effective delivery of large genes to the retina by dual AAV vectors. EMBO Mol. Med. 2014, 6, 194–211. [Google Scholar] [CrossRef] [PubMed]
- Colella, P.; Trapani, I.; Cesi, G.; Sommella, A.; Manfredi, A.; Puppo, A.; Iodice, C.; Rossi, S.; Simonelli, F.; Giunti, M.; et al. Efficient gene delivery to the cone-enriched pig retina by dual AAV vectors. Gene Ther. 2014, 21, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, A.; Tornabene, P.; Tiberi, P.; Minopoli, R.; Manfredi, A.; Mutarelli, M.; Auricchio, A. Triple Vectors Expand AAV Transfer Capacity in the Retina. Mol. Ther. 2018, 26, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Baye, L.M.; Patrinostro, X.; Swaminathan, S.; Beck, J.S.; Zhang, Y.; Stone, E.M.; Sheffield, V.C.; Slusarski, D.C. The N-terminal region of centrosomal protein 290 (CEP290) restores vision in a zebrafish model of human blindness. Hum. Mol. Genet. 2011, 20, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Sanjurjo-Soriano, C.; Kalatzis, V. Guiding Lights in Genome Editing for Inherited Retinal Disorders: Implications for Gene and Cell Therapy. Neural Plast. 2018, 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F., III. ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Wang, X.; Hu, X.; Liu, Z.; Liu, J.; Zhou, H.; Shen, X.; Wei, Y.; Huang, Z.; Ying, W.; et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017, 27, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Schierling, B.; Dannemann, N.; Gabsalilow, L.; Wende, W.; Cathomen, T.; Pingoud, A. A novel zinc-finger nuclease platform with a sequence-specific cleavage module. Nucleic Acids Res. 2012, 40, 2623–2638. [Google Scholar] [CrossRef] [PubMed]
- Greenwald, D.L.; Cashman, S.M.; Kumar-Singh, R. Engineered Zinc Finger Nuclease–Mediated Homologous Recombination of the Human Rhodopsin Gene. Investig. Opthalmol. Vis. Sci. 2010, 51, 6374–6380. [Google Scholar] [CrossRef] [PubMed]
- Low, B.E.; Krebs, M.P.; Joung, J.K.; Tsai, S.Q.; Nishina, P.M.; Wiles, M.V. Correction of the Crb1rd8 Allele and Retinal Phenotype in C57BL/6N Mice Via TALEN-Mediated Homology-Directed Repair. Investig. Opthalmol. Vis. Sci. 2014, 55, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera RM, C.; Asokan, A. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Hung, S.S.C.; Chrysostomou, V.; Li, F.; Lim, J.K.H.; Wang, J.-H.; Powell, J.E.; Tu, L.; Daniszewski, M.; Lo, C.; Wong, R.C.; et al. AAV-Mediated CRISPR/Cas Gene Editing of Retinal Cells In Vivo. Investig. Opthalmol. Vis. Sci. 2016, 57, 3470. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Cong, L. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; Amoasii, L.; Mireault, A.A.; McAnally, J.R.; Li, H.; Sanchez-Ortiz, E.; Olson, E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 2016, 351, 400–403. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.-S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, Y.; Kweon, J.; Kim, H.S.; Bae, S.; Kim, J.S. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014, 24, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kweon, J.; Kim, A.; Chon, J.K.; Yoo, J.Y.; Kim, H.J.; Kim, S.; Lee, C.; Jeong, E.; Chung, E.; et al. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 2013, 31, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Peddle, C.F.; MacLaren, R.E. The Application of CRISPR/Cas9 for the Treatment of Retinal Diseases. Yale J. Boil. Med. 2017, 90, 533–541. [Google Scholar]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Jo, D.H.; Koo, T.; Cho, C.S.; Kim, J.H.; Kim, J.-S.; Kim, J.H. Long-Term Effects of In Vivo Genome Editing in the Mouse Retina Using Campylobacter jejuni Cas9 Expressed via Adeno-Associated Virus. Mol. Ther. 2019, 27, 130–136. [Google Scholar] [CrossRef]
- Li, F.; Hung, S.S.; Khalid, M.K.N.M.; Wang, J.-H.; Chrysostomou, V.; Wong, V.H.; Singh, V.; Wing, K.; Tu, L.; Bender, J.A.; et al. Utility of self-destructing CRISPR/Cas constructs for targeted gene editing in the retina. Hum. Gene Ther. 2019. [Google Scholar] [CrossRef]
- Zelinka, C.P.; Sotolongo-Lopez, M.; Fadool, J.M. Targeted disruption of the endogenous zebrafish rhodopsin locus as models of rapid rod photoreceptor degeneration. Mol. Vis. 2018, 24, 587–602. [Google Scholar]
- Xia, C.-H.; Ferguson, I.; Li, M.; Kim, A.; Onishi, A.; Li, L.; Su, B.; Gong, X. Essential function of NHE8 in mouse retina demonstrated by AAV-mediated CRISPR/Cas9 knockdown. Exp. Eye Res. 2018, 176, 29–39. [Google Scholar] [CrossRef]
- Moreno, A.M.; Fu, X.; Zhu, J.; Katrekar, D.; Shih, Y.-R.V.; Marlett, J.; Cabotaje, J.; Tat, J.; Naughton, J.; Lisowski, L.; et al. In Situ Gene Therapy via AAV-CRISPR-Cas9-Mediated Targeted Gene Regulation. Mol. Ther. 2018, 26, 1818–1827. [Google Scholar] [CrossRef]
- Giannelli, S.G.; Luoni, M.; Castoldi, V.; Massimino, L.; Cabassi, T.; Angeloni, D.; Broccoli, V. Cas9/sgRNA selective targeting of the P23H Rhodopsin mutant allele for treating retinitis pigmentosa by intravitreal AAV9.PHP.B-based delivery. Hum. Mol. Genet. 2018, 27, 761–779. [Google Scholar] [CrossRef]
- Burnight, E.R.; Gupta, M.; Wiley, L.A.; Anfinson, K.R.; Tran, A.; Triboulet, R.; Hoffmann, J.M.; Klaahsen, D.L.; Andorf, J.L.; Jiao, C.; et al. Using CRISPR-Cas9 to Generate Gene-Corrected Autologous iPSCs for the Treatment of Inherited Retinal Degeneration. Mol. Ther. 2017, 25, 1999–2013. [Google Scholar] [CrossRef]
- Kim, E.; Koo, T.; Park, S.W.; Kim, D.; Kim, K.; Cho, H.-Y.; Song, D.W.; Lee, K.J.; Jung, M.H.; Kim, S.; et al. In vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017, 8, 14500. [Google Scholar] [CrossRef]
- Latella, M.C.; Di Salvo, M.T.; Cocchiarella, F.; Benati, D.; Grisendi, G.; Comitato, A.; Marigo, V.; Recchia, A. In vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. Nucleic Acids 2016, 5, e389. [Google Scholar] [CrossRef]
- Bassuk, A.G.; Zheng, A.; Li, Y.; Tsang, S.H.; Mahajan, V.B. Precision Medicine: Genetic Repair of Retinitis Pigmentosa in Patient-Derived Stem Cells. Sci. Rep. 2016, 6, 19969. [Google Scholar] [CrossRef]
- Wu, W.-H.; Tsai, Y.-T.; Justus, S.; Lee, T.-T.; Zhang, L.; Lin, C.-S.; Bassuk, A.G.; Mahajan, V.B.; Tsang, S.H. CRISPR Repair Reveals Causative Mutation in a Preclinical Model of Retinitis Pigmentosa. Mol. Ther. 2016, 24, 1388–1394. [Google Scholar] [CrossRef]
- Zhong, H.; Chen, Y.; Li, Y.; Chen, R.; Mardon, G. CRISPR-engineered mosaicism rapidly reveals that loss of Kcnj13 function in mice mimics human disease phenotypes. Sci. Rep. 2015, 5, 8366. [Google Scholar] [CrossRef]
- Swiech, L.; Heidenreich, M.; Banerjee, A.; Habib, N.; Li, Y.; Trombetta, J.; Zhang, F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015, 33, 102–106. [Google Scholar] [CrossRef]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 2014, 3, 04766. [Google Scholar] [CrossRef]
- Kim, K.; Park, S.W.; Kim, J.H.; Lee, S.H.; Kim, D.; Koo, T.; Kim, K.-E.; Kim, J.H.; Kim, J.-S. Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res. 2017, 27, 419–426. [Google Scholar] [CrossRef]
- Suzuki, K.; Tsunekawa, Y.; Hernandez-Benitez, R.; Wu, J.; Zhu, J.; Kim, E.J.; Hatanaka, F.; Yamamoto, M.; Araoka, T.; Li, Z.; et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 2016, 540, 144–149. [Google Scholar] [CrossRef]
- Diakatou, M.; Manes, G.; Bocquet, B.; Meunier, I.; Kalatzis, V. Genome Editing as a Treatment for the Most Prevalent Causative Genes of Autosomal Dominant Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 20, 2542. [Google Scholar] [CrossRef]
- Garanto, A.; Van Beersum, S.E.C.; Peters, T.A.; Roepman, R.; Cremers, F.P.M.; Collin, R.W.J. Unexpected CEP290 mRNA Splicing in a Humanized Knock-In Mouse Model for Leber Congenital Amaurosis. PLoS ONE 2013, 8, e79369. [Google Scholar] [CrossRef]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef]
- Duebel, J.; Marazova, K.; Sahel, J.A. Optogenetics. Curr. Opin. Ophthalmol. 2015, 26, 226–232. [Google Scholar] [CrossRef]
- Simunovic, M.; Shen, W.; Lin, J.; Protti, D.; Lisowski, L.; Gillies, M. Optogenetic approaches to vision restoration. Exp. Eye Res. 2019, 178, 15–26. [Google Scholar] [CrossRef]
- Deisseroth, K.; Feng, G.; Majewska, A.K.; Miesenböck, G.; Ting, A.; Schnitzer, M.J. Next-Generation Optical Technologies for Illuminating Genetically Targeted Brain Circuits. J. Neurosci. 2006, 26, 10380–10386. [Google Scholar] [CrossRef]
- Yue, L.; Weiland, J.D.; Roska, B.; Humayun, M.S. Retinal stimulation strategies to restore vision: Fundamentals and systems. Prog. Retin. Eye Res. 2016, 53, 21–47. [Google Scholar] [CrossRef]
- Zhang, F.; Vierock, J.; Yizhar, O.; Fenno, L.E.; Tsunoda, S.; Kianianmomeni, A.; Prigge, M.; Berndt, A.; Cushman, J.; Polle, J.; et al. The Microbial Opsin Family of Optogenetic Tools. Cell 2011, 147, 1446–1457. [Google Scholar] [CrossRef]
- Bernstein, J.G.; Boyden, E.S. Optogenetic tools for analyzing the neural circuits of behavior. Trends Cogn. Sci. 2011, 15, 592–600. [Google Scholar] [CrossRef]
- Ahnelt, P.K.; Kolb, H. The mammalian photoreceptor mosaic-adaptive design. Prog. Retin. Eye Res. 2000, 19, 711–777. [Google Scholar] [CrossRef]
- Gaub, B.M.; Berry, M.H.; Holt, A.E.; Isacoff, E.Y.; Flannery, J.G. Optogenetic Vision Restoration Using Rhodopsin for Enhanced Sensitivity. Mol. Ther. 2015, 23, 1562–1571. [Google Scholar] [CrossRef]
- Sengupta, A.; Chaffiol, A.; Macé, E.; Caplette, R.; Desrosiers, M.; Lampič, M.; Forster, V.; Marre, O.; Lin, J.Y.; Sahel, J.; et al. Red-shifted channelrhodopsin stimulation restores light responses in blind mice, macaque retina, and human retina. EMBO Mol. Med. 2016, 8, 1248–1264. [Google Scholar] [CrossRef]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef]
- Hammond, S.M.; Wood, M.J. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef]
- Garanto, A.; Collin, R.W.J. Applications of antisense oligonucleotides for the treatment of inherited retinal diseases. Curr. Opin. Ophthalmol. 2017, 28, 1–266. [Google Scholar]
- Bacchi, N.; Casarosa, S.; Denti, M.A. Splicing-Correcting Therapeutic Approaches for Retinal Dystrophies: Where Endogenous Gene Regulation and Specificity Matter. Investig. Opthalmol. Vis. Sci. 2014, 55, 3285–3294. [Google Scholar] [CrossRef]
- Chan, J.H.; Lim, S.; Wong, W.F. Antisense oligonucleotides: from design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Vulin, A.; Fougerousse, F.; Leturcq, F.; Kaplan, J.-C.; García, L.; Danos, O. Rescue of Dystrophic Muscle Through U7 snRNA-Mediated Exon Skipping. Science 2004, 306, 1796–1799. [Google Scholar] [CrossRef]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar]
- Vitravene Study Group. Safety of intravitreous fomivirsen for treatment of cytomegalovirus retinitis in patients with AIDS. Am. J. Ophthalmol. 2002, 133, 484–498. [Google Scholar]
- Vitravene Study Group. Randomized dose-comparison studies of intravitreous fomivirsen for treatment of cytomegalovirus retinitis that has reactivated or is persistently active despite other therapies in patients with AIDS. Am. J. Ophthalmol. 2002, 133, 475–483. [Google Scholar]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.J.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) Gene Are a Frequent Cause of Leber Congenital Amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef]
- Collin, R.W.; Hollander, A.I.D.; Van Der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol. Ther. Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.-J.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef]
- Duijkers, L.; van den Born, L.; Neidhardt, J.; Bax, N.; Pierrache, L.; Klevering, B.; Garanto, A. Antisense Oligonucleotide-Based Splicing Correction in Individuals with Leber Congenital Amaurosis due to Compound Heterozygosity for the c.2991+1655A>G Mutation in CEP290. Int. J. Mol. Sci. 2018, 19, 753. [Google Scholar] [CrossRef]
- Dulla, K.; Aguila, M.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Schulkens, I.; Collin, R.W. Splice-Modulating Oligonucleotide QR-110 Restores CEP290 mRNA and Function in Human c.2991+1655A>G LCA10 Models. Mol. Ther. Nucleic Acids 2018, 12, 730–740. [Google Scholar] [CrossRef]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Pfeifer, W.L. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef]
- Barny, I.; Perrault, I.; Michel, C.; Goudin, N.; Defoort-Dhellemmes, S.; Ghazi, I.; Kaplan, J.; Rozet, J.-M.; Gerard, X. AON-Mediated Exon Skipping to Bypass Protein Truncation in Retinal Dystrophies Due to the Recurrent CEP290 c.4723A > T Mutation. Fact or Fiction? Genes 2019, 10, 368. [Google Scholar] [CrossRef]
- Bonifert, T.; Menendez, I.G.; Battke, F.; Theurer, Y.; Synofzik, M.; Schöls, L.; Wissinger, B. Antisense Oligonucleotide Mediated Splice Correction of a Deep Intronic Mutation in OPA1. Mol. Ther. Nucleic Acids 2016, 5, e390. [Google Scholar] [CrossRef]
- Garanto, A.; Van Der Velde-Visser, S.D.; Cremers, F.P.M.; Collin, R.W.J. Antisense Oligonucleotide-Based Splice Correction of a Deep-Intronic Mutation in CHM Underlying Choroideremia. In Results and Problems in Cell Differentiation; Springer: Cham, Switzerland, 2018; pp. 83–89. [Google Scholar]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; Peters, T.A.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther. Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; Born, L.I.V.D.; Khan, M.I.; Cornelis, S.S.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W. Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-Intronic ABCA4 Variant c.4539+2001G>A in Stargardt Disease. Genes 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed]
- Tanner, G.; Glaus, E.; Barthelmes, D.; Ader, M.; Fleischhauer, J.; Pagani, F.; Berger, W.; Neidhardt, J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum. Mutat. 2009, 30, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Glaus, E.; Barthelmes, D.; Fliegauf, M.; Gaspar, H.; Nürnberg, G.; Nürnberg, P.; Omran, H.; Berger, W.; Neidhardt, J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011, 32, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene Therapeutic Approach Using Mutation-adapted U1 snRNA to Correct a RPGR Splice Defect in Patient-derived Cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef]
- Schmid, F.; Hiller, T.; Korner, G.; Glaus, E.; Berger, W.; Neidhardt, J. A Gene Therapeutic Approach to Correct Splice Defects with Modified U1 and U6 snRNPs. Hum. Gene Ther. 2013, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Abad, X.; Vera, M.; Jung, S.P.; Oswald, E.; Romero, I.; Amin, V.; Fortes, P.; Gunderson, S.I. Requirements for gene silencing mediated by U1 snRNA binding to a target sequence. Nucleic Acids Res. 2008, 36, 2338–2352. [Google Scholar] [CrossRef]
- Berger, A.; Lorain, S.; Joséphine, C.; Desrosiers, M.; Peccate, C.; Voit, T.; Garcia, L.; Sahel, J.-A.; Bemelmans, A.-P. Repair of Rhodopsin mRNA by Spliceosome-Mediated RNA Trans. -Splicing: A New Approach for Autosomal Dominant Retinitis Pigmentosa. Mol. Ther. 2015, 23, 918–930. [Google Scholar] [CrossRef]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Boil. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef]
- Li, N.; Zheng, J.; Li, H.; Deng, J.; Hu, M.; Wu, H.; Li, W.; Li, F.; Lan, X.; Lu, J.; et al. Identification of chimeric TSNAX–DISC1 resulting from intergenic splicing in endometrial carcinoma through high-throughput RNA sequencing. Carcinogenesis 2014, 35, 2687–2697. [Google Scholar] [CrossRef]
- Guerra, E.; Trerotola, M.; Arciprete, R.D.; Bonasera, V.; Palombo, B.; El-Sewedy, T.; Ciccimarra, T.; Crescenzi, C.; Lorenzini, F.; Rossi, C.; et al. A Bicistronic CYCLIN D1-TROP2 mRNA Chimera Demonstrates a Novel Oncogenic Mechanism in Human Cancer. Cancer Res. 2008, 68, 8113–8121. [Google Scholar] [CrossRef] [PubMed]
- Hirano, M.; Noda, T. Genomic organization of the mouse Msh4 gene producing bicistronic, chimeric and antisense mRNA. Gene 2004, 342, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.J.; McDougald, D.S.; Fisher, K.J.; Bennicelli, J.L.; Mitchell, L.G.; Bennett, J. Spliceosome-Mediated Pre-mRNA trans-Splicing Can Repair CEP290 mRNA. Mol. Ther. Nucleic Acids 2018, 12, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Saurabh, S.; Vidyarthi, A.S.; Prasad, D. RNA interference: Concept to reality in crop improvement. Planta 2014, 239, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Adema, C.M.; Lane, T. A computational study of off-target effects of RNA interference. Nucleic Acids Res. 2005, 33, 1834–1847. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, N.-K.; Lee, J.; Lee, H.; Hong, H.K.; Kim, H.; Lee, J.B.; Woo, S.J.; Park, K.H.; Kim, H. Therapeutic effects of a novel siRNA-based anti-VEGF (siVEGF) nanoball for the treatment of choroidal neovascularization. Nanoscale 2017, 9, 15461–15469. [Google Scholar] [CrossRef] [PubMed]
- Yau, E.H.; Butler, M.C.; Sullivan, J.M. A Cellular High-Throughput Screening Approach for Therapeutic trans-Cleaving Ribozymes and RNAi against Arbitrary mRNA Disease Targets. Exp. Eye Res. 2016, 151, 236–255. [Google Scholar] [CrossRef] [PubMed]
- Lagos-Quintana, M.; Rauhut, R.; Meyer, J.; Borkhardt, A.; Tuschl, T. New microRNAs from mouse and human. RNA 2003, 9, 175–179. [Google Scholar] [CrossRef]
- Zuzic, M.; Arias, J.E.R.; Wohl, S.G.; Busskamp, V. Retinal miRNA Functions in Health and Disease. Genes 2019, 10, 377. [Google Scholar] [CrossRef]
- Mangos, M.M.; Min, K.-L.; Viazovkina, E.; Galarneau, A.; Elzagheid, M.I.; Parniak, M.A.; Damha, M.J. Efficient RNase H-Directed Cleavage of RNA Promoted by Antisense DNA or 2‘F-ANA Constructs Containing Acyclic Nucleotide Inserts. J. Am. Chem. Soc. 2003, 125, 654–661. [Google Scholar] [CrossRef]
- Honcharenko, D.; Barman, J.; Varghese, O.P. Comparison of the RNase H Cleavage Kinetics and Blood Serum Stability of theNorth-Conformationally Constrained and 2‘-Alkoxy Modified Oligonucleotides†. Biochemistry 2007, 46, 5635–5646. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-Specific Inhibition of Rhodopsin With an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investig. Opthalmol. Vis. Sci. 2015, 56, 6362–6375. [Google Scholar] [CrossRef] [PubMed]
- Naessens, S.; Ruysschaert, L.; Lefever, S.; Coppieters, F.; De Baere, E. Antisense Oligonucleotide-Based Downregulation of the G56R Pathogenic Variant Causing NR2E3-Associated Autosomal Dominant Retinitis Pigmentosa. Genes 2019, 10, 363. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR–Cas systems. Nat. Rev. Genet. 2017, 15, 169–182. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Lander, E.S. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef]
- Stafforst, T.; Schneider, M.F. An RNA-Deaminase Conjugate Selectively Repairs Point Mutations. Angew. Chem. Int. Ed. 2012, 51, 11166–11169. [Google Scholar] [CrossRef] [PubMed]
- Wettengel, J.; Reautschnig, P.; Geisler, S.; Kahle, P.J.; Stafforst, T. Harnessing human ADAR2 for RNA repair - Recoding a PINK1 mutation rescues mitophagy. Nucleic Acids Res. 2017, 45, 2797–2808. [Google Scholar] [CrossRef]
- Vighi, E.; Trifunović, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; Heuvel, A.V.D.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef]
- Nagel-Wolfrum, K.; Möller, F.; Penner, I.; Baasov, T.; Wolfrum, U. Targeting Nonsense Mutations in Diseases with Translational Read-Through-Inducing Drugs (TRIDs). BioDrugs 2016, 30, 49–74. [Google Scholar] [CrossRef]
- Schwarz, N.; Carr, A.J.; Lane, A.; Moeller, F.; Chen, L.L.; Aguila, M.; Nagel-Wolfrum, K. Translational read-through of the RP2 Arg120stop mutation in patient iPSC-derived retinal pigment epithelium cells. Hum. Mol. Genet. 2015, 24, 972–986. [Google Scholar] [CrossRef] [PubMed]
- Guérin, K.; Gregory-Evans, C.; Hodges, M.D.J.; Moosajee, M.; Mackay, D.; Gregory-Evans, K.; Flannery, J.G.; Mackay, D. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp. Eye Res. 2008, 87, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Overlack, N.; Möller, F.; Belakhov, V.; Van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Rebibo-Sabbah, A.; Overlack, N.; Nudelman, I.; Belakhov, V.; Baasov, T.; Ben-Yosef, T.; Wolfrum, U.; Nagel-Wolfrum, K. Beneficial Read-Through of a USH1C Nonsense Mutation by Designed Aminoglycoside NB30 in the Retina. Investig. Opthalmol. Vis. Sci. 2010, 51, 6671–6680. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Glikin, D.; Smolkin, B.; Hainrichson, M.; Belakhov, V.; Baasov, T. Repairing faulty genes by aminoglycosides: Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorganic Med. Chem. 2010, 18, 3735–3746. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Rebibo-Sabbah, A.; Cherniavsky, M.; Belakhov, V.; Hainrichson, M.; Chen, F.; Schacht, J.; Pilch, D.S.; Ben-Yosef, T.; Baasov, T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Rebibo-Sabbah, A.; Shallom-Shezifi, D.; Hainrichson, M.; Stahl, I.; Ben-Yosef, T.; Baasov, T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorganic Med. Chem. Lett. 2006, 16, 6310–6315. [Google Scholar] [CrossRef]
- Rebibo-Sabbah, A.; Nudelman, I.; Ahmed, Z.M.; Baasov, T.; Ben-Yosef, T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Qual. Life Res. 2007, 122, 373–381. [Google Scholar] [CrossRef]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef]
- Shimizu-Motohashi, Y.; Komaki, H.; Motohashi, N.; Takeda, S.; Yokota, T.; Aoki, Y. Restoring Dystrophin Expression in Duchenne Muscular Dystrophy: Current Status of Therapeutic Approaches. J. Pers. Med. 2019, 9, 1. [Google Scholar] [CrossRef]
- Sermet-Gaudelus, I.; De Boeck, K.; Casimir, G.J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; et al. Ataluren (PTC124) Induces Cystic Fibrosis Transmembrane Conductance Regulator Protein Expression and Activity in Children with Nonsense Mutation Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic Aminoglycosides Efficiently Suppress Cystic Fibrosis Transmembrane Conductance Regulator Nonsense Mutations and Are Enhanced by Ivacaftor. Am. J. Respir. Cell Mol. Boil. 2014, 50, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Moosajee, M.; Tracey-White, D.; Smart, M.; Weetall, M.; Torriano, S.; Kalatzis, V.; Da Cruz, L.; Coffey, P.; Webster, A.R.; Welch, E. Functional rescue of REP1 following treatment with PTC124 and novel derivative PTC-414 in human choroideremia fibroblasts and the nonsense-mediated zebrafish model. Hum. Mol. Genet. 2016, 25, 3416–3431. [Google Scholar] [CrossRef] [PubMed]
- Torriano, S.; Erkilic, N.; Baux, D.; Cereso, N.; De Luca, V.; Meunier, I.; Moosajee, M.; Roux, A.-F.; Hamel, C.P.; Kalatzis, V. The effect of PTC124 on choroideremia fibroblasts and iPSC-derived RPE raises considerations for therapy. Sci. Rep. 2018, 8, 8234. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, H.; Mitsios, A.; Smart, M.; Skinner, J.; Welch, A.A.; Kalatzis, V.; Coffey, P.J.; Dubis, A.M.; Webster, A.R.; Moosajee, M. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum. Mol. Genet. 2019, 28, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Madni, A.; Sarfraz, M.; Rehman, M.; Ahmad, M.; Akhtar, N.; Ahmad, S.; Tahir, N.; Ijaz, S.; Al-Kassas, R.; Löbenberg, R.; et al. Liposomal Drug Delivery: A Versatile Platform for Challenging Clinical Applications. J. Pharm. Pharm. Sci. 2014, 17, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Cheetham, M.E. Targeting the Proteostasis Network in Rhodopsin Retinitis Pigmentosa. Adv. Exp. Med. Biol. 2016, 854, 479–484. [Google Scholar] [PubMed]
- Faber, S.; Roepman, R. Balancing the Photoreceptor Proteome: Proteostasis Network Therapeutics for Inherited Retinal Disease. Genes 2019, 10, 557. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Aguila, M.; McCulley, C.H.; Bevilacqua, D.; Mendes, H.F.; Athanasiou, D.; Novoselov, S.S.; Kanuga, N.; Munro, P.M.; Coffey, P.J.; et al. The heat-shock response co-inducer arimoclomol protects against retinal degeneration in rhodopsin retinitis pigmentosa. Cell Death Dis. 2014, 5, e1236. [Google Scholar] [CrossRef]
- Athanasiou, D.; Aguila, M.; Bellingham, J.; Li, W.; McCulley, C.; Reeves, P.J.; Cheetham, M.E. The molecular and cellular basis of rhodopsin retinitis pigmentosa reveals potential strategies for therapy. Prog. Retin Eye Res. 2018, 62, 1–23. [Google Scholar] [CrossRef]
- Mendes, H.F.; Cheetham, M.E. Pharmacological manipulation of gain-of-function and dominant-negative mechanisms in rhodopsin retinitis pigmentosa. Hum. Mol. Genet. 2008, 17, 3043–3054. [Google Scholar] [CrossRef] [PubMed]
- Bogéa, T.H.; Wen, R.H.; Moritz, O.L. Light Induces Ultrastructural Changes in Rod Outer and Inner Segments, Including Autophagy, in a Transgenic Xenopus laevis P23H Rhodopsin Model of Retinitis Pigmentosa. Investig. Opthalmol. Vis. Sci. 2015, 56, 7947–7955. [Google Scholar] [CrossRef] [PubMed]
- Sakami, S.; Maeda, T.; Bereta, G.; Okano, K.; Golczak, M.; Sumaroka, A.; Roman, A.J.; Cideciyan, A.V.; Jacobson, S.G.; Palczewski, K. Probing Mechanisms of Photoreceptor Degeneration in a New Mouse Model of the Common Form of Autosomal Dominant Retinitis Pigmentosa due to P23H Opsin Mutations. J. Boil. Chem. 2011, 286, 10551–10567. [Google Scholar] [CrossRef] [PubMed]
- Lax, P.; Pinilla, I.; Cuenca, N.; Fernández-Sánchez, L.; Martín-Nieto, J. Tauroursodeoxycholic Acid Prevents Retinal Degeneration in Transgenic P23H Rats. Investig. Opthalmol. Vis. Sci. 2011, 52, 4998–5008. [Google Scholar]
- Tao, Y.; Dong, X.; Lu, X.; Qu, Y.; Wang, C.; Peng, G.; Zhang, J. Subcutaneous delivery of tauroursodeoxycholic acid rescues the cone photoreceptors in degenerative retina: A promising therapeutic molecule for retinopathy. Biomed. Pharmacother. 2019, 117, 109021. [Google Scholar] [CrossRef] [PubMed]
- Lobysheva, E.; Taylor, C.; Marshall, G.; Kisselev, O. Tauroursodeoxycholic acid binds to the G-protein site on light activated rhodopsin. Exp. Eye Res. 2018, 170, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Remondelli, P.; Renna, M. The Endoplasmic Reticulum Unfolded Protein Response in Neurodegenerative Disorders and Its Potential Therapeutic Significance. Front. Mol. Neurosci. 2017, 10, 187. [Google Scholar] [CrossRef]
- Tolone, A.; Belhadj, S.; Rentsch, A.; Schwede, F.; Paquet-Durand, F. The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes 2019, 10, 453. [Google Scholar] [CrossRef] [PubMed]
- Palczewski, K. Retinoids for Treatment of Retinal Diseases. Trends Pharmacol. Sci. 2010, 31, 284–295. [Google Scholar] [CrossRef]
- Scholl, H.P.N.; Moore, A.T.; Koenekoop, R.K.; Wen, Y.; Fishman, G.A.; Born, L.I.V.D.; Bittner, A.; Bowles, K.; Fletcher, E.C.; Collison, F.T.; et al. Safety and Proof-of-Concept Study of Oral QLT091001 in Retinitis Pigmentosa Due to Inherited Deficiencies of Retinal Pigment Epithelial 65 Protein (RPE65) or Lecithin:Retinol Acyltransferase (LRAT). PLoS ONE 2015, 10, e0143846. [Google Scholar] [CrossRef]
- Patel, A.; Cholkar, K.; Agrahari, V.; Mitra, A.K. Ocular drug delivery systems: An overview. World J. Gastrointest. Pharmacol. Ther. 2013, 2, 47–64. [Google Scholar] [CrossRef] [PubMed]
- Bordet, T.; Behar-Cohen, F. Ocular gene therapies in clinical practice: Viral vectors and nonviral alternatives. Drug Discov. Today 2019, 24, 1685–1693. [Google Scholar] [CrossRef]
- Conley, S.M.; Cai, X.; Naash, M.I. Non-Viral Ocular Gene Therapy: Assessment and Future Directions. Curr. Opin. Mol. Ther. 2008, 10, 456–463. [Google Scholar] [PubMed]
- Chiang, B.; Jung, J.H.; Prausnitz, M.R. The suprachoroidal space as a route of administration to the posterior segment of the eye. Adv. Drug Deliv. Rev. 2018, 126, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, G.A.; Shalaev, E.; Karami, T.K.; Cunningham, J.; Slater, N.K.H.; Rivers, H.M. Pharmaceutical Development of AAV-Based Gene Therapy Products for the Eye. Pharm. Res. 2018, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Olsen, T.W.; Feng, X.; Wabner, K.; Conston, S.R.; Sierra, D.H.; Folden, D.V.; Smith, M.E.; Cameron, J.D. Cannulation of the Suprachoroidal Space: A Novel Drug Delivery Methodology to the Posterior Segment. Am. J. Ophthalmol. 2006, 142, 777–787.e2. [Google Scholar] [CrossRef] [PubMed]
- Einmahl, S.; Savoldelli, M.; D’Hermies, F.; Tabatabay, C.; Gurny, R.; Behar-Cohen, F. Evaluation of a novel biomaterial in the suprachoroidal space of the rabbit eye. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1533–1539. [Google Scholar]
- Mandelcorn, E.D.; Kitchens, J.W.; Fijalkowski, N.; Moshfeghi, D.M. Active Aspiration of Suprachoroidal Hemorrhage Using a Guarded Needle. Ophthalmic Surg. Lasers Imaging Retin. 2014, 45, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.A.; Do, D.; Noronha, G.; Kissner, J.M.; Srivastava, S.K.; Nguyen, Q.D. Suprachoroidal Corticosteroid Administration: A Novel Route for Local Treatment of Noninfectious Uveitis. Transl. Vis. Sci. Technol. 2016, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Gote, V.; Sikder, S.; Sicotte, J.; Pal, D. Ocular Drug Delivery: Present Innovations and Future Challenges. J. Pharmacol. Exp. Ther. 2019, 370, 602–624. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Ashtari, M.; Wellman, J.; Marshall, K.A.; Cyckowski, L.L.; Chung, D.C.; McCague, S.; Pierce, E.A.; Chen, Y.; Bennicelli, J.L.; et al. AAV2 Gene Therapy Readministration in Three Adults with Congenital Blindness. Sci. Transl. Med. 2012, 4, 120ra15. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I.; Puppo, A.; Auricchio, A. Vector platforms for gene therapy of inherited retinopathies. Prog. Retin. Eye Res. 2014, 43, 108–128. [Google Scholar] [CrossRef] [PubMed]
- Stieger, K.; Lhériteau, E.; Moullier, P.; Rolling, F. AAV-Mediated Gene Therapy for Retinal Disorders in Large Animal Models. ILAR J. 2009, 50, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Petrs-Silva, H.; Dinculescu, A.; Li, Q.; Deng, W.T.; Pang, J.J.; Min, S.H.; Agbandje-McKenna, M. Novel properties of tyrosine-mutant AAV2 vectors in the mouse retina. Mol. Ther. 2011, 19, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Li, W.; Li, X.; Zheng, Q.; Dai, X.; Zhou, X.; Boye, S.L.; Hauswirth, W.W.; Qu, J.; Pang, J.-J. Self-complementary AAV5 Vector Facilitates Quicker Transgene Expression in Photoreceptor and Retinal Pigment Epithelial Cells of Normal Mouse. Exp. Eye Res. 2010, 90, 546–554. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghosh, A.; Yue, Y.; Duan, D. Efficient transgene reconstitution with hybrid dual AAV vectors carrying the minimized bridging sequences. Hum. Gene Ther. 2011, 22, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Trapani, I. Dual AAV Vectors for Stargardt Disease. Methods Mol. Biol. 2018, 1715, 153–175. [Google Scholar] [PubMed]
- Trapani, I. Adeno-Associated Viral Vectors as a Tool for Large Gene Delivery to the Retina. Genes 2019, 10, 287. [Google Scholar] [CrossRef]
- Patel, A.; Zhao, J.; Duan, D.; Lai, Y. Design of AAV Vectors for Delivery of Large or Multiple Transgenes. Adv. Struct. Saf. Stud. 2019, 1950, 19–33. [Google Scholar]
- Auricchio, A.; Kobinger, G.; Anand, V.; Hildinger, M.; O’Connor, E.; Maguire, A.M.; Wilson, J.M.; Bennett, J. Exchange of surface proteins impacts on viral vector cellular specificity and transduction characteristics: The retina as a model. Hum. Mol. Genet. 2001, 10, 3075–3081. [Google Scholar] [CrossRef]
- Martin, K.R.; Klein, R.L.; Quigley, H.A. Gene delivery to the eye using adeno-associated viral vectors. Methods 2002, 28, 267–275. [Google Scholar] [CrossRef]
- Dalkara, D.; Byrne, L.C.; Klimczak, R.R.; Visel, M.; Yin, L.; Merigan, W.H.; Flannery, J.G.; Schaffer, D.V. In Vivo-Directed Evolution of a New Adeno-Associated Virus for Therapeutic Outer Retinal Gene Delivery from the Vitreous. Sci. Transl. Med. 2013, 5, 189ra76. [Google Scholar] [CrossRef] [PubMed]
- Cronin, T.; Vandenberghe, L.H.; Hantz, P.; Juttner, J.; Reimann, A.; Kacsó, Á.E.; Huckfeldt, R.M.; Busskamp, V.; Kohler, H.; Lagali, P.S.; et al. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol. Med. 2014, 6, 1175–1190. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.S.; Lee, V.; Wei, Z.; Song, J.Y.; Casal, G.; Cronin, T.; Willett, K.; Huckfeldt, R.; Morgan, J.I.; Aleman, T.S.; et al. Evaluation of Dose and Safety of AAV7m8 and AAV8BP2 in the Non-Human Primate Retina. Hum. Gene Ther. 2017, 28, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, L.S.; Xiao, R.; Wassmer, S.J.; Langsdorf, A.; Zinn, E.; Pacouret, S.; Shah, S.; Comander, J.I.; Kim, L.A.; Lim, L.; et al. Synthetic Adeno-Associated Viral Vector Efficiently Targets Mouse and Nonhuman Primate Retina In Vivo. Hum. Gene Ther. 2018, 29, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, K.P.; Lee, E.S.; Schaffer, D.V.; Flannery, J.G. Gene delivery to the retina using lentiviral vectors. Adv. Exp. Med. Biol. 2006, 572, 255–266. [Google Scholar]
- Han, Z.; Conley, S.M.; Naash, M.I. Gene Therapy for Stargardt Disease Associated with ABCA4 Gene. Adv. Exp. Med. Biol. 2014, 801, 719–724. [Google Scholar]
- Bergelson, J.M.; Modlin, J.F.; Wieland-Alter, W.; Cunningham, J.A.; Crowell, R.L.; Finberg, R.W. Clinical Coxsackievirus B Isolates Differ from Laboratory Strains in Their Interaction with Two Cell Surface Receptors. J. Infect. Dis. 1997, 175, 697–700. [Google Scholar] [CrossRef]
- Cashman, S.M.; Sadowski, S.L.; Morris, D.J.; Frederick, J.; Kumar-Singh, R. Intercellular Trafficking of Adenovirus-Delivered HSV VP22 from the Retinal Pigment Epithelium to the Photoreceptors—Implications for Gene Therapy. Mol. Ther. 2002, 6, 813–823. [Google Scholar] [CrossRef]
- Mallam, J.N.; Hurwitz, M.Y.; Mahoney, T.; Che´vez-Barrios, P.; Hurwitz, R.L. Efficient gene transfer into retinal cells using adenoviral vectors: Dependence on receptor expression. Investig. Opthalmol. Vis. Sci. 2004, 45, 1680–1687. [Google Scholar] [CrossRef]
- Kumar-Singh, R.; Farber, D.B. Encapsidated adenovirus mini-chromosome-mediated delivery of genes to the retina: Application to the rescue of photoreceptor degeneration. Hum. Mol. Genet. 1998, 7, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a Common Receptor for Coxsackie B Viruses and Adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Andrieu-Soler, C.; Bejjani, R.-A.; De Bizemont, T.; Normand, N.; Benezra, D.; Behar-Cohen, F. Ocular gene therapy: A review of nonviral strategies. Mol. Vis. 2006, 12, 1334–1347. [Google Scholar] [PubMed]
- Foroozandeh, P.; Aziz, A.A. Insight into Cellular Uptake and Intracellular Trafficking of Nanoparticles. Nanoscale Res. Lett. 2018, 13, 339. [Google Scholar] [CrossRef] [PubMed]
- Trigueros, S.; Domènech, E.B.; Toulis, V.; Marfany, G. In Vitro Gene Delivery in Retinal Pigment Epithelium Cells by Plasmid DNA-Wrapped Gold Nanoparticles. Genes 2019, 10, 289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Gold Nanoparticles: Recent Advances in the Biomedical Applications. Cell Biophys. 2015, 72, 771–775. [Google Scholar] [CrossRef]
- Peeters, L.; Sanders, N.N.; Braeckmans, K.; Boussery, K.; Van De Voorde, J.; De Smedt, S.C.; Demeester, J. Vitreous: A barrier to nonviral ocular gene therapy. Investig. Opthalmol. Vis. Sci. 2005, 46, 3553–3561. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; McLenachan, S.; Edel, M.; Da Cruz, L.; Coffey, P.; Mackey, D. iPS Cells for Modelling and Treatment of Retinal Diseases. J. Clin. Med. 2014, 3, 1511–1541. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Lamba, D.A.; Gust, J.; Reh, T.A. Transplantation of human embryonic stem cells derived photoreceptors restores some visual function in Crx deficient mice. Cell Stem Cell 2009, 4, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.A.; Barber, A.C.; Rizzi, M.; Hippert, C.; Xue, T.; West, E.L.; Duran, Y.; Smith, A.J.; Chuang, J.Z.; Azam, S.A.; et al. Restoration of vision after transplantation of photoreceptors. Nature 2012, 485, 99–103. [Google Scholar] [CrossRef] [PubMed]
- West, E.L.; Gonzalez-Cordero, A.; Hippert, C.; Osakada, F.; Martinez-Barbera, J.P.; Pearson, R.A.; Ali, R.R. Defining the integration capacity of embryonic stem cell-derived photoreceptor precursors. Stem Cells 2012, 30, 1424–1435. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.C.; Hippert, C.; Duran, Y.; West, E.L.; Bainbridge, J.W.; Warre-Cornish, K.; Pearson, R.A. Repair of the degenerate retina by photoreceptor transplantation. Proc. Natl. Acad Sci. USA 2013, 110, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.A.; Cordero, A.G.; West, E.L.; Ribeiro, J.R.; Aghaizu, N.; Goh, D.; Sampson, R.D.; Georgiadis, A.; Waldron, P.V.; Duran, Y.; et al. Donor and host photoreceptors engage in material transfer following transplantation of post-mitotic photoreceptor precursors. Nat. Commun. 2016, 7, 13029. [Google Scholar] [CrossRef] [PubMed]
- Santos-Ferreira, T.; Llonch, S.; Börsch, O.; Postel, K.; Haas, J.; Ader, M. Retinal transplantation of photoreceptors results in donor–host cytoplasmic exchange. Nat. Commun. 2016, 7, 13028. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, H.W.; Wang, L.; Li, S.Y.; Zhao, C.J.; Hao, J.; Li, Q.Y.; Zhao, T.T.; Wu, W.; Wang, Y.; et al. Human embryonic stem cell-derived retinal pigment epithelium transplants as a potential treatment for wet age-related macular degeneration. Cell Discov. 2018, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.D.; Regillo, C.D.; Lam, B.L.; Eliott, D.; Rosenfeld, P.J.; Gregori, N.Z.; Hubschman, J.-P.; Davis, J.L.; Heilwell, G.; Spirn, M.; et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt’s macular dystrophy: Follow-up of two open-label phase 1/2 studies. Lancet 2015, 385, 509–516. [Google Scholar] [CrossRef]
- Song, W.K.; Park, K.-M.; Kim, H.-J.; Lee, J.H.; Choi, J.; Chong, S.Y.; Shim, S.H.; Del Priore, L.V.; Lanza, R. Treatment of Macular Degeneration Using Embryonic Stem Cell-Derived Retinal Pigment Epithelium: Preliminary Results in Asian Patients. Stem Cell Rep. 2015, 4, 860–872. [Google Scholar] [CrossRef] [PubMed]
- Mandai, M.; Watanabe, A.; Kurimoto, Y.; Hirami, Y.; Morinaga, C.; Daimon, T.; Fujihara, M.; Akimaru, H.; Sakai, N.; Shibata, Y.; et al. Autologous Induced Stem-Cell–Derived Retinal Cells for Macular Degeneration. N. Engl. J. Med. 2017, 376, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Zrenner, E. Will Retinal Implants Restore Vision? Science 2002, 295, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Bloch, E.; Luo, Y.; Da Cruz, L. Advances in retinal prosthesis systems. Ther. Adv. Ophthalmol. 2019, 11, 2515841418817501. [Google Scholar] [CrossRef] [PubMed]
- da Cruz, L.; Dorn, J.D.; Humayun, M.S.; Dagnelie, G.; Handa, J.; Barale, P.O.; Salzmann, J. Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis System Clinical Trial. Ophthalmology 2016, 123, 2248–2254. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gene/Condition | Therapeutic Molecule | Clinical Trial Identifier | Status |
|---|---|---|---|
| Gene Augmentation | |||
| ABCA4 | SAR422459 | NCT01367444 | Recruiting |
| SAR422459 | NCT01736592 | Enrolling by invitation | |
| CHM | AAV2-hCHM | NCT02341807 | Active, not recruiting |
| AAV2-REP1 | NCT03496012 | Recruiting | |
| AAV2-REP1 | NCT03507686 | Recruiting | |
| AAV2-REP1 | NCT02407678 | Active, not recruiting | |
| rAAV2.REP1 | NCT02671539 | Active, not recruiting | |
| rAAV2.REP1 | NCT02077361 | Completed | |
| rAAV2.REP1 | NCT01461213 | Completed | |
| AAV2-REP1 | NCT03584165 | Enrolling by invitation | |
| AAV2-REP1 | NCT02553135 | Completed | |
| CNGA3 | AGTC-402 | NCT02935517 | Recruiting |
| AAV2/8-hG1.7p.coCNGA3 | NCT03758404 | Recruiting | |
| rAAV.hCNGA3 | NCT02610582 | Active, not recruiting | |
| CNGB3 | rAAV2tYF-PR1.7-hCNGB3 | NCT02599922 | Recruiting |
| AAV2/8-hCARp.hCNGB3 | NCT03001310 | Recruiting | |
| AAV-CNGB3 | NCT03278873 | Recruiting | |
| MERTK | rAAV2-VMD2-hMERTK | NCT01482195 | Recruiting |
| MYO7A | UshStat | NCT02065011 | Enrolling by invitation |
| PDE6B | AAV2/5-hPDE6B | NCT03328130 | Recruiting |
| RPE65 | AAV OPTIREP | NCT02946879 | Recruiting |
| rAAV2-CBSB-hRPE65 | NCT00481546 | Active, not recruiting | |
| AAV2-hRPE65v2- | NCT00516477 | Active, not recruiting | |
| AAV2-hRPE65v2 | NCT00999609 | Active, not recruiting | |
| AAV2-hRPE65v2 | NCT01208389 | Active, not recruiting | |
| AAV2-hRPE65v2 | NCT03602820 | Active, not recruiting | |
| tgAAG76 (rAAV 2/2.hRPE65p.hRPE65) | NCT00643747 | Completed | |
| rAAV2-CB-hRPE65 | NCT00749957 | Completed | |
| rAAV2-hRPE65 | NCT00821340 | Completed | |
| rAAV2/4.hRPE65 | NCT01496040 | Completed | |
| AAV RPE65 | NCT02781480 | Completed | |
| RLBP1 | CPK850 | NCT03374657 | Recruiting |
| RPGR | AAV8-RPGR | NCT03116113 | Recruiting |
| AAV2/5-hRKp.RPGR | NCT03252847 | Recruiting | |
| rAAV2tYF-GRK1-RPGR | NCT03316560 | Recruiting | |
| RS1 | AAV8-scRS/IRBPhRS | NCT02317887 | Recruiting |
| rAAV2tYF-CB-hRS1 | NCT02416622 | Active, not recruiting | |
| Genome editing | |||
| CEP290 | EDIT-101 (AGN-151587) | NCT03872479 | Recruiting |
| Optogenetics | |||
| Advanced RP | RST-001 | NCT02556736 | Recruiting |
| Non-syndromic RP Retinitis Pigmentosa | GS030-DP | NCT03326336 | Recruiting |
| Gene | Therapeutic Molecule | Clinical Trial Identifier | Status |
|---|---|---|---|
| CEP290 | QR-110 | NCT03140969 | Active, not recruiting |
| QR-110 | NCT03913130 | Recruiting | |
| QR-110 | NCT03913143 | Recruiting | |
| USH2A | QR-421a | NCT03780257 | Recruiting |
| Gene | Therapeutic Molecule | Clinical Trial Identifier | Status |
|---|---|---|---|
| ABCA4 | ALK-001 | NCT02402660 | Recruiting |
| Zimura | NCT03364153 | Active, not recruiting | |
| Emuxustat | NCT03772665 | Recruiting | |
| Emuxustat | NCT03033108 | Completed | |
| RPE65 | QLT091001 | NCT01014052 | Completed |
| QLT091001 | NCT01521793 | Completed | |
| QLT091001 | NCT01543906 * | Completed | |
| RS1 | Dorzolamide 2% TID or brinzolamide 1% TID | NCT02331173 | Completed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. https://doi.org/10.3390/genes10090654
Vázquez-Domínguez I, Garanto A, Collin RWJ. Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges. Genes. 2019; 10(9):654. https://doi.org/10.3390/genes10090654
Chicago/Turabian StyleVázquez-Domínguez, Irene, Alejandro Garanto, and Rob W. J. Collin. 2019. "Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges" Genes 10, no. 9: 654. https://doi.org/10.3390/genes10090654
APA StyleVázquez-Domínguez, I., Garanto, A., & Collin, R. W. J. (2019). Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges. Genes, 10(9), 654. https://doi.org/10.3390/genes10090654