Profiling the Functional Diversity of Termite Mound Soil Bacteria as Revealed by Shotgun Sequencing



Abstract
1. Introduction
2. Materials and Methods
2.1. Study Sites and Soil Sampling
2.2. Soil Analysis of Termite Mound Soils and Their Comparative Surrounding Soil Samples
2.3. Metagenomic DNA Extraction and Sequencing
2.4. Metagenome Annotation And Data Analysis
3. Results
3.1. Metagenome Sequencing and Sequence Processing
3.2. Functional Analysis Associated with Termite Mound Soils and Their Surrounding Soil Samples
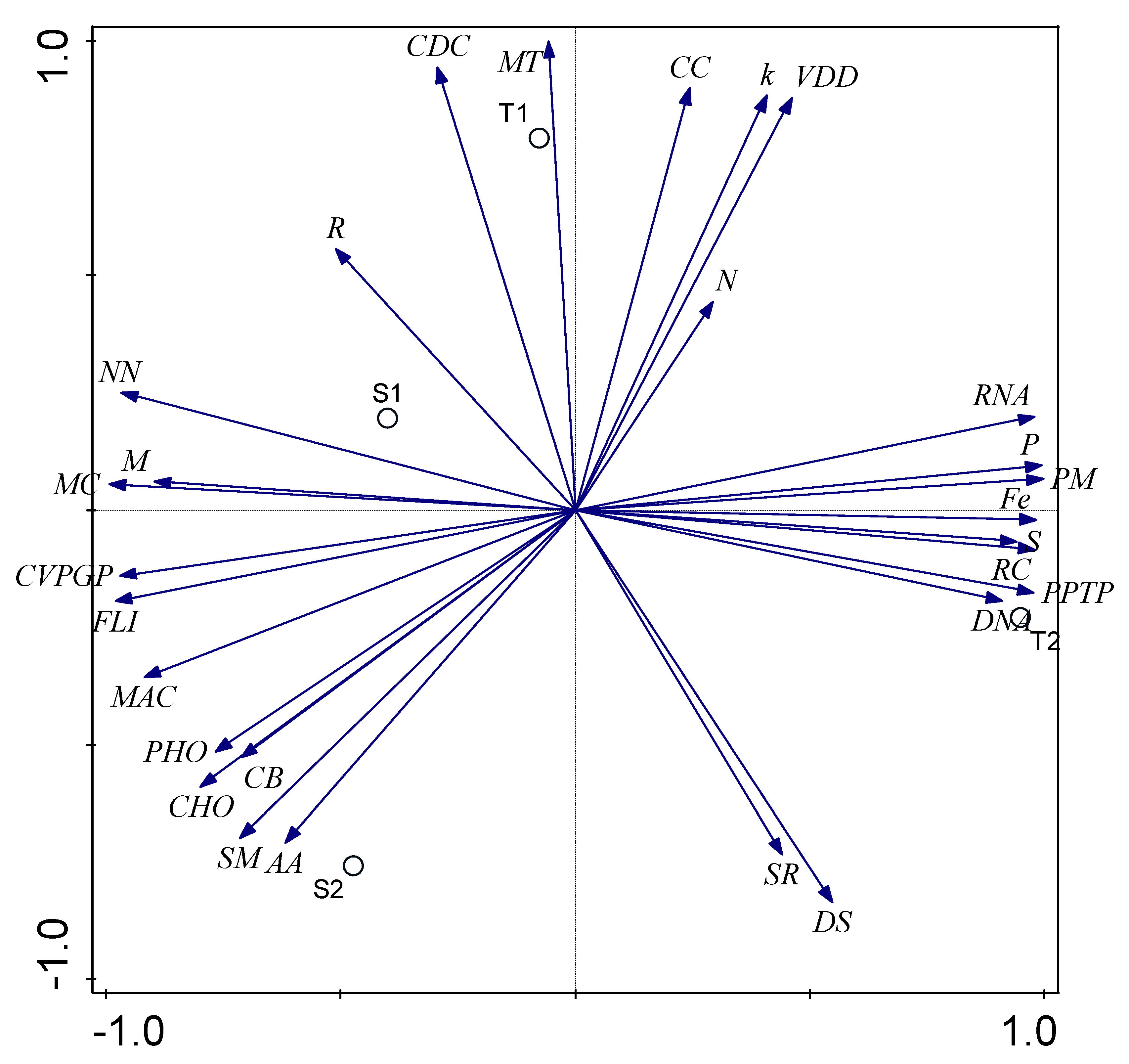
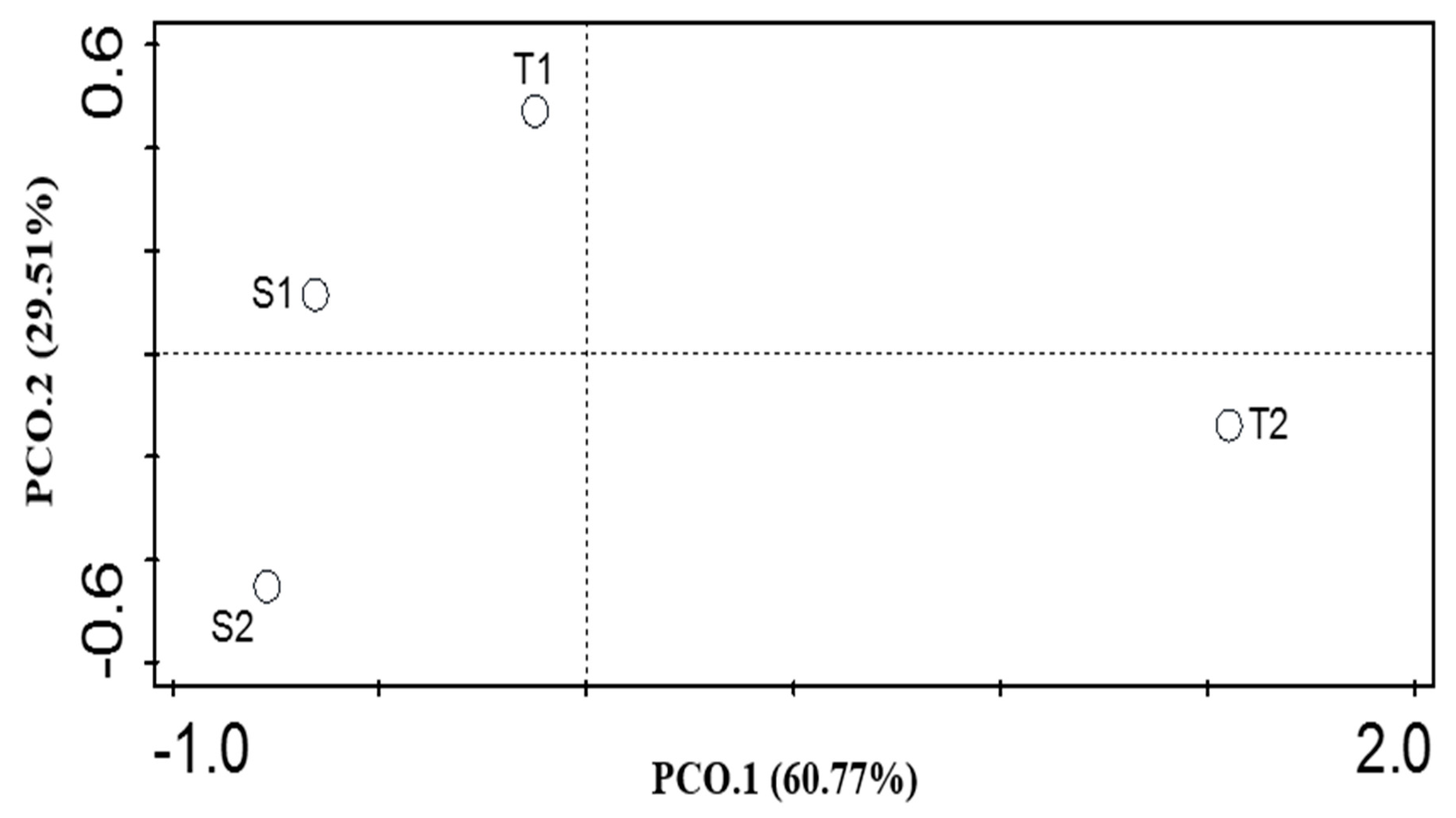
3.3. α and β Diversity of the Functional Categories of both Soil Samples
3.4. Physiochemical Characterization of the Termite Mound Soils and Their Surrounding Soil Samples
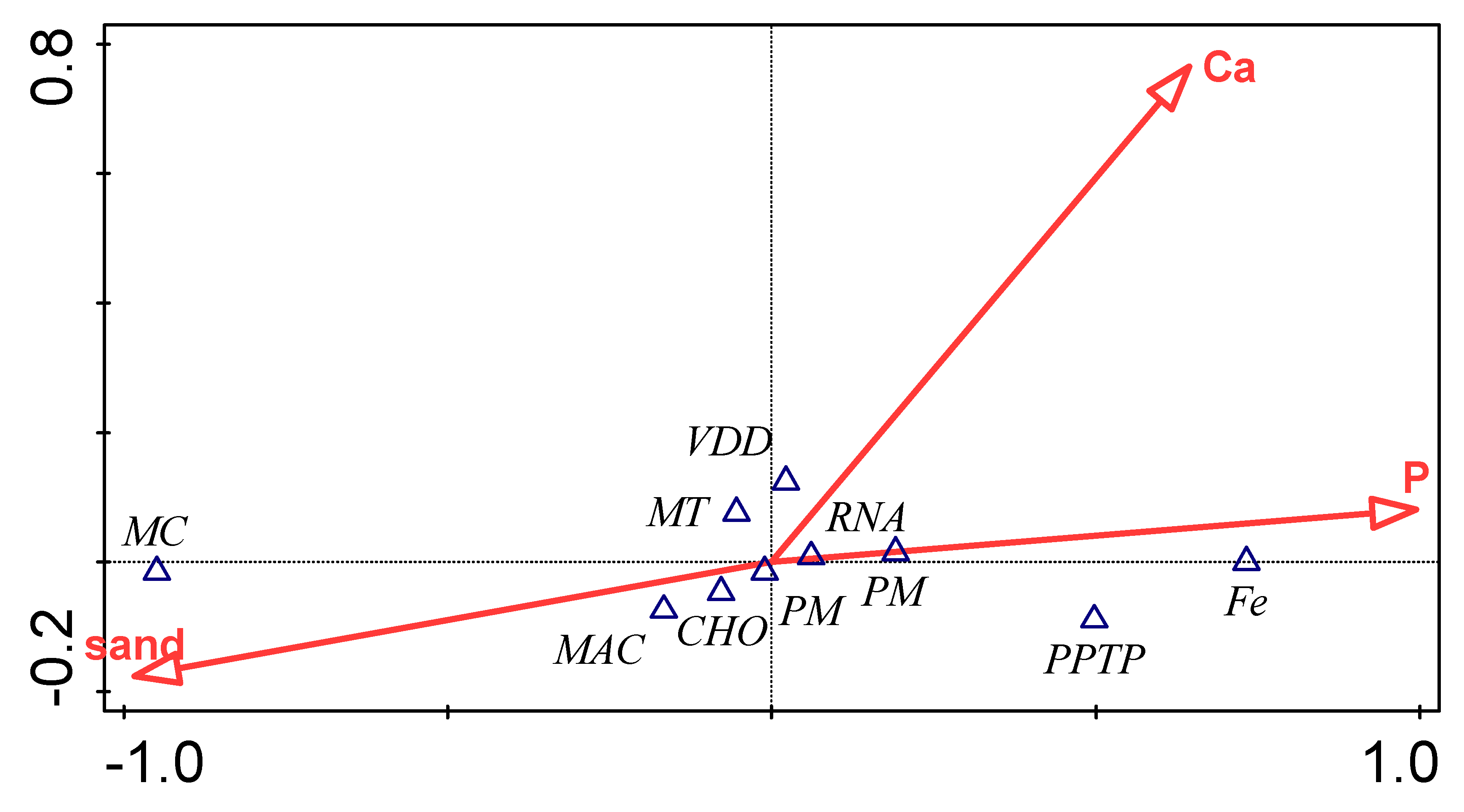
3.5. Influence of Environmental Factors on Bacterial Functional Category
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fall, S.; Hamelin, J.; Ndiaye, F.; Assigbetse, K.; Aragno, M.; Chotte, J.L.; Brauman, A. Differences between bacterial communities in the gut of a soil-feeding termite (cubitermes niokoloensis) and its mounds. Appl. Environ. Microbiol. 2007, 73, 5199–5208. [Google Scholar] [CrossRef] [PubMed]
- Manjula, A.; Pushpanathan, M.; Sathyavathi, S.; Gunasekaran, P.; Rajendhran, J. Comparative analysis of microbial diversity in termite gut and termite nest using ion sequencing. Curr. Microbiol. 2016, 72, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Krishanti, N.P.R.A.; Zulfina, D.; Wikantyoso, B.; Zulfitri, A.; Yusuf, S. Antimicrobial production by an actinomycetes isolated from the termite nest. J. Trop. Life Sci. 2018, 8. [Google Scholar] [CrossRef]
- Sujada, N.; Sungthong, R.; Lumyong, S. Termite nests as an abundant source of cultivable actinobacteria for biotechnological purposes. Microbes Environ. 2014, 29, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Duponnois, R.; Kisa, M.; Assigbetse, K.; Prin, Y.; Thioulouse, J.; Issartel, M.; Moulin, P.; Lepage, M. Fluorescent pseudomonads occuring in macrotermes subhyalinus mound structures decrease cd toxicity and improve its accumulation in sorghum plants. Sci. Total Environ. 2006, 370, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Nauer, P.A.; Hutley, L.B.; Arndt, S.K. Termite mounds mitigate half of termite methane emissions. Proc. Natl. Acad. Sci. USA 2018, 115, 13306–13311. [Google Scholar] [CrossRef]
- Enagbonma, B.J.; Babalola, O.O. Potentials of termite mound soil bacteria in ecosystem engineering for sustainable agriculture. Ann. Microbiol. 2019, 69, 211–219. [Google Scholar] [CrossRef]
- Enagbonma, B.J.; Babalola, O.O. Environmental sustainability: A review of termite mound soil material and its bacteria. Sustainability 2019, 11, 3847. [Google Scholar] [CrossRef]
- Harry, M.; Jusseaume, N.; Gambier, B.; Garnier-Sillam, E. Use of rapd markers for the study of microbial community similarity from termite mounds and tropical soils. Soil Biol. Biochem. 2001, 33, 417–427. [Google Scholar] [CrossRef]
- Dhembare, A. Physico-chemical properties of termite mound soil. Arch. Appl. Sci. Res. 2013, 5, 123–126. [Google Scholar]
- Deke, A.L.; Adugna, W.T.; Fite, A.T. Soil physic-chemical properties in termite mounds and adjacent control soil in miyo and yabello districts of borana zone, southern ethiopia. Am. J. Agric. For. 2016, 4, 69–74. [Google Scholar]
- Bhatia, C.R. Role of microbial diversity for soil, health and plant nutrition. In Molecular Mechanisms of Plant and Microbe Coexistence; Nautiyal, C.S., Dion, P., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 53–74. [Google Scholar]
- Spain, A.V.; Gordon, V.; Reddell, P.; Correll, R. Ectomycorrhizal fungal spores in the mounds of tropical australian termites (isoptera). Eur. J. Soil Biol. 2004, 40, 9–14. [Google Scholar] [CrossRef]
- Kumar, P.; Tilak, M.; Sivakumar, K.; Saranya, K. Studies on the assessment of major nutrients and microbial population of termite mound soil. Int. J. For. Crop Improv. 2018, 9, 13–17. [Google Scholar] [CrossRef]
- Zhu, L.-X.; Xiao, Q.; Shen, Y.-F.; Li, S.-Q. Microbial functional diversity responses to 2 years since biochar application in silt-loam soils on the loess plateau. Ecotoxicol. Environ. Saf. 2017, 144, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.; Ghosh, D.; Sarkar, D. Biogeochemical cycling bacteria and nutrient dynamics in waste stabilization pond system. In Wastewater Management through Aquaculture; Jana, B.B., Mandal, R.N., Jayasankar, P., Eds.; Springer: Singapore, 2018; pp. 29–52. [Google Scholar]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33–45. [Google Scholar] [CrossRef]
- Torsvik, V.; Øvreås, L. Microbial diversity and function in soil: From genes to ecosystems. Curr. Opin. Microbiol. 2002, 5, 240–245. [Google Scholar] [CrossRef]
- Vera-Gargallo, B.; Ventosa, A. Metagenomic insights into the phylogenetic and metabolic diversity of the prokaryotic community dwelling in hypersaline soils from the odiel saltmarshes (sw spain). Genes 2018, 9, 152. [Google Scholar] [CrossRef]
- Chouvenc, T.; Efstathion, C.A.; Elliott, M.L.; Su, N.-Y. Extended disease resistance emerging from the faecal nest of a subterranean termite. Proc. R. Soc. Biol. Sci. 2013, 280, 20131885. [Google Scholar] [CrossRef]
- Chen, L.; Luo, Y.; Xu, J.; Yu, Z.; Zhang, K.; Brookes, P.C. Assessment of bacterial communities and predictive functional profiling in soils subjected to short-term fumigation-incubation. Micro. Ecol. 2016, 72, 240–251. [Google Scholar] [CrossRef]
- Ranjan, R.; Rani, A.; Metwally, A.; McGee, H.S.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16s amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Kettler, T.; Doran, J.W.; Gilbert, T. Simplified method for soil particle-size determination to accompany soil-quality analyses. Soil Sci. Soc. Am. J. 2001, 65, 849–852. [Google Scholar] [CrossRef]
- Muwawa, E.M.; Makonde, H.M.; Budambula, N.; Osiemo, Z.L. Chemical properties associated with guts, soil and nest materials of odontotermes and macrotermes species from kenya. J. Biol. Environ. Sci. 2010, 4, 253–263. [Google Scholar]
- Shi, J.-Y.; Yuan, X.-F.; Lin, H.-R.; Yang, Y.-Q.; Li, Z.-Y. Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant fritillaria thunbergii. Int. J. Mol. Sci. 2011, 12, 3770–3785. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics rast server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. Blat—The blast-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The m5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Hammer, Ř.; Harper, D.; Ryan, P. Past: Paleontological statistics software package for education and data analysis–palaeontol. Palaeontol. Electron. 2001, 4, 9–15. [Google Scholar]
- Clarke, K.; Green, R. Statistical design and analysis for a “biological effects” study. Mar. Ecol. Prog. Ser. 1988, 46, 213–226. [Google Scholar] [CrossRef]
- Khomtchouk, B.B.; Hennessy, J.R.; Wahlestedt, C. Shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE 2017, 12, e0176334. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.-Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Ibekwe, A.M.; Leddy, M.; Yang, C.-H.; Crowley, D.E. Assimilable organic carbon (aoc) in soil water extracts using vibrio harveyi bb721 and its implication for microbial biomass. PLoS ONE 2012, 7, e28519. [Google Scholar] [CrossRef] [PubMed]
- Gianoulis, T.A.; Raes, J.; Patel, P.V.; Bjornson, R.; Korbel, J.O.; Letunic, I.; Yamada, T.; Paccanaro, A.; Jensen, L.J.; Snyder, M. Quantifying environmental adaptation of metabolic pathways in metagenomics. Proc. Natl. Acad. Sci. USA 2009, 8, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Torsvik, V.; Øvreås, L. Microbial diversity, life strategies, and adaptation to life in extreme soils. In Microbiology of Extreme Soils; Springer: Berlin/Heidelberg, Germany, 2008; pp. 15–43. [Google Scholar]
- Lindsay, E.A.; Colloff, M.J.; Gibb, N.L.; Wakelin, S.A. The abundance of microbial functional genes in grassy woodlands is influenced more by soil nutrient enrichment than by recent weed invasion or livestock exclusion. Appl. Environ. Microbiol. 2010, 76, 5547–5555. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, L.E.; Barbosa, O. Metagenomic analysis exploring taxonomic and functional diversity of soil microbial communities in chilean vineyards and surrounding native forests. PeerJ 2017, 5, e3098. [Google Scholar] [CrossRef] [PubMed]
- Uroz, S.; Ioannidis, P.; Lengelle, J.; Cébron, A.; Morin, E.; Buée, M.; Martin, F. Functional assays and metagenomic analyses reveals differences between the microbial communities inhabiting the soil horizons of a norway spruce plantation. PLoS ONE 2013, 8, e55929. [Google Scholar] [CrossRef] [PubMed]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497. [Google Scholar] [CrossRef]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a ph gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Cassman, N.; de Hollander, M.; Mendes, L.W.; Korevaar, H.; Geerts, R.H.; van Veen, J.A.; Kuramae, E.E. Impact of long-term n, p, k, and npk fertilization on the composition and potential functions of the bacterial community in grassland soil. FEMS Microbiol. Ecol. 2014, 90, 195–205. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T1 | T2 | S1 | S2 | p-Value | |
|---|---|---|---|---|---|
| Shannon_H | 2.86 ± 0.16 | 2.87 ± 0.16 | 2.85 ± 0.16 | 2.84 ± 0.16 | 0.99 |
| Evenness_e^H/S | 0.62 ± 0.06 | 0.63 ± 0.06 | 0.62 ± 0.07 | 0.61 ± 0.07 |
| Soil Property | T1 | T2 | S1 | S2 |
|---|---|---|---|---|
| Sand (%) | 65.00 ± 8.29a | 47.75 ± 23.60b | 72.00 ± 17.66c | 76.50 ± 3.00d |
| Silt (%) | 9.00 ± 2.94a | 19.75 ± 11.38a | 11.75 ± 12.87a | 10.25 ± 0.96a |
| Clay (%) | 26.00 ± 6.27a | 33.25 ± 13.52a | 16.25 ± 4.50b | 13.25 ± 3.20c |
| K (mg/L) | 393.50 ± 120.33a | 427.50 ± 57.93a | 216.75 ± 48.40b | 184.50 ± 27.72c |
| Ca (mg/L) | 1879.50 ± 587.38a | 2237.75 ± 318.91a | 1493.50 ± 456.59a | 1108.50 ± 160.48b |
| Mg (mg/L) | 575.00 ± 262.32a | 622.25 ± 60.84a | 349.75 ± 159.70a | 330.25 ± 138.75a |
| pH | 5.10 ± 0.33a | 4.48 ± 0.46a | 5.80 ± 0.32b | 5.38 ± 0.39c |
| N (%) | 0.09 ± 0.03a | 0.10 ± 0.03b | 0.59 ± 0.47c | 0.25 ± 0.04d |
| P (mg/L) | 0.25 ± 0.50a | 0.75 ± 0.50a | 0.00 ± 0.00a | 0.00 ± 0.00a |
| OC (%) | 0.31 ± 0.42a | 0.10 ± 0.00a | 0.11 ± 0.0a | 0.11 ± 0.01a |
| Environmental Variable | Explains % | Contribution % | Pseudo-F | P |
|---|---|---|---|---|
| P | 92.8 | 92.8 | 25.8 | 0.09 |
| Ca | 5.1 | 5.1 | 2.4 | 0.87 |
| Sand | 2.1 | 2.1 | <0.1 | 1. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enagbonma, B.J.; Aremu, B.R.; Babalola, O.O. Profiling the Functional Diversity of Termite Mound Soil Bacteria as Revealed by Shotgun Sequencing. Genes 2019, 10, 637. https://doi.org/10.3390/genes10090637
Enagbonma BJ, Aremu BR, Babalola OO. Profiling the Functional Diversity of Termite Mound Soil Bacteria as Revealed by Shotgun Sequencing. Genes. 2019; 10(9):637. https://doi.org/10.3390/genes10090637
Chicago/Turabian StyleEnagbonma, Ben Jesuorsemwen, Bukola Rhoda Aremu, and Olubukola Oluranti Babalola. 2019. "Profiling the Functional Diversity of Termite Mound Soil Bacteria as Revealed by Shotgun Sequencing" Genes 10, no. 9: 637. https://doi.org/10.3390/genes10090637
APA StyleEnagbonma, B. J., Aremu, B. R., & Babalola, O. O. (2019). Profiling the Functional Diversity of Termite Mound Soil Bacteria as Revealed by Shotgun Sequencing. Genes, 10(9), 637. https://doi.org/10.3390/genes10090637