DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut

 ,
,  ,
,
Abstract
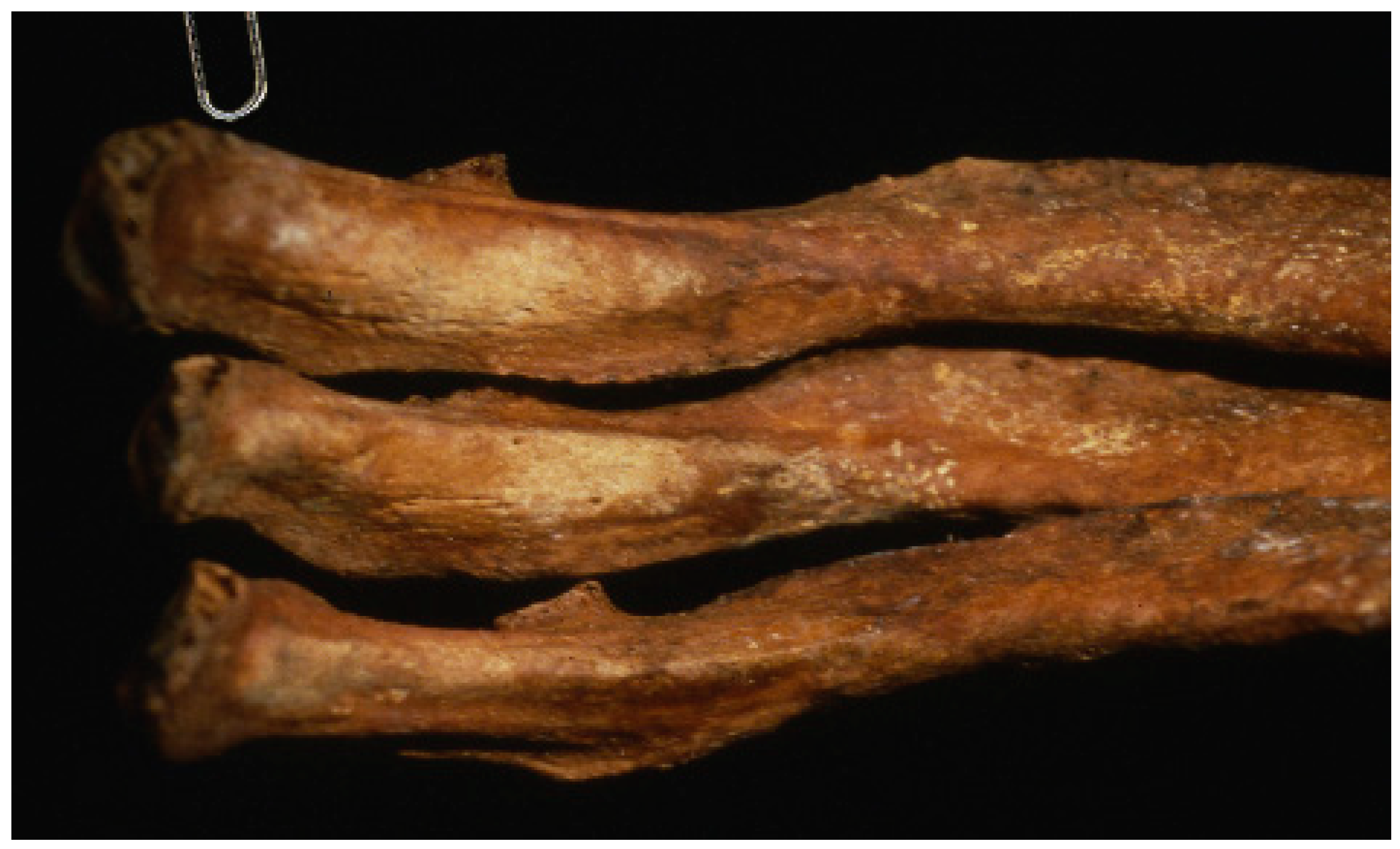
1. Introduction
2. Materials and Methods
2.1. Contamination Prevention
2.2. DNA Extraction
2.3. DNA Repair and Purification
2.4. DNA Quantification
2.5. Precision ID Ancestry SNP Panel
2.6. Y-STR Typing and Y-haplogroup Prediction
3. Results
3.1. DNA Quantification
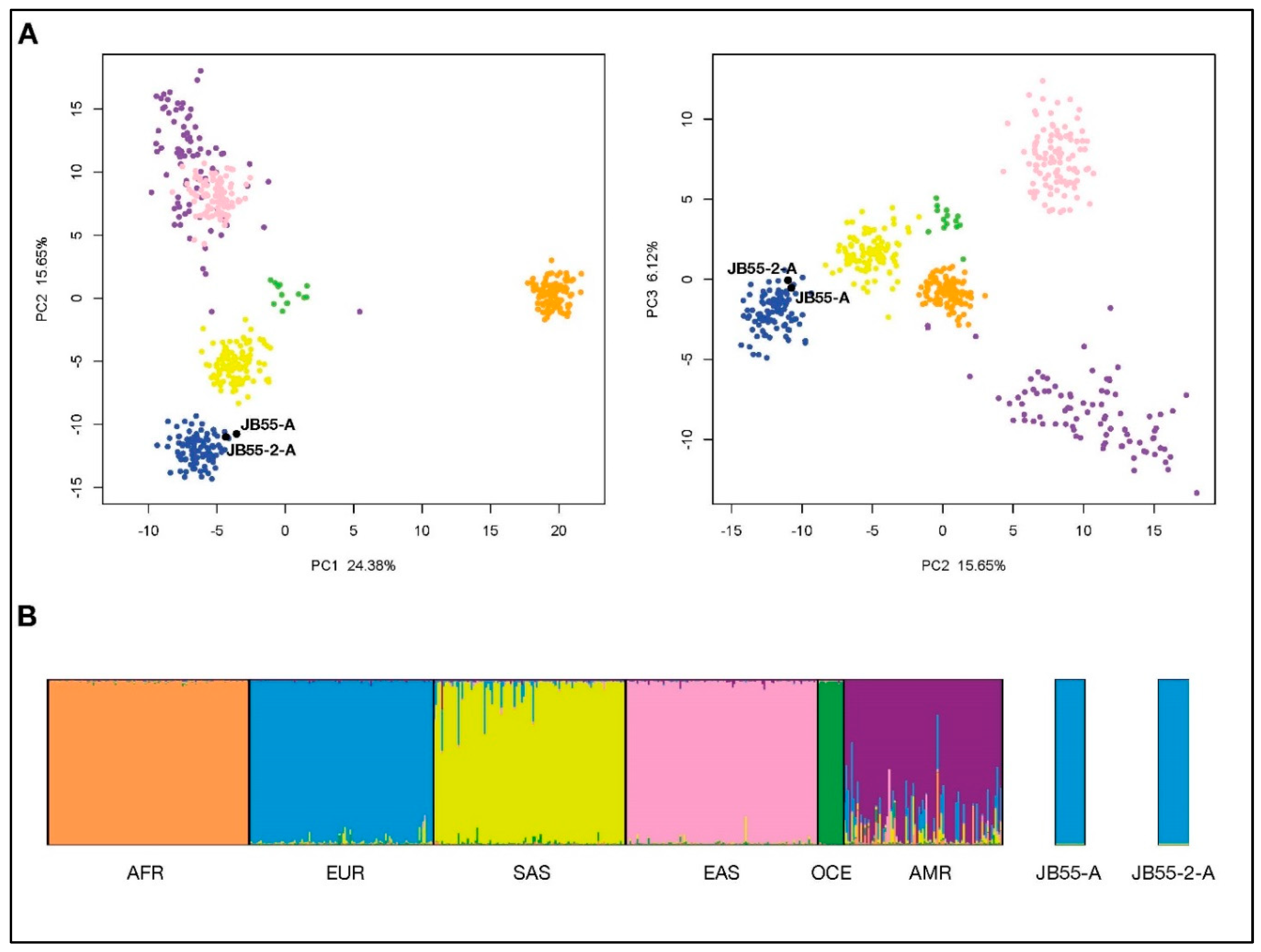
3.2. Ancestry Estimation from Precision ID Ancestry SNP Panel
3.3. Y-STR Analysis
3.3.1. Yfiler Y-STR Typing
3.3.2. Y Haplogroup Estimation and Surname Prediction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Illumina Sequencing of Yfiler Amplification Product
References
- Bellantoni, N.F.; Sledzik, P.S.; Poirier, D.A. Rescue, Research, and Reburial: Walton Family Cemetery, Griswold, Connecticut. In Remembrance: Archaeology and Death; Bergin & Garvey: Westport, CT, USA, 1997; pp. 132–154. [Google Scholar]
- Kelley, M.A.; Micozzi, M.S. Rib Lesions in Chronic Pulmonary Tuberculosis. Am. J. Phys. Anthropol. 1984, 65, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.A.; Eisenberg, L.E. Blastomycosis and Tuberculosis in Early American Indians: A Biocultural View. Midcont. J. Archaeol. 1987, 12, 89–116. [Google Scholar]
- Sledzik, P.S.; Bellantoni, N. Brief Communication: Bioarcheological and Biocultural Evidence for the New England Vampire Folk Belief. Am. J. Phys. Anthropol. 1994, 94, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A. The Great New England Vampire Panic. Smithsonian 2012, 43, 58–66. [Google Scholar]
- Bell, M.E. Food for the Dead: On the Trail of New England’s Vampires; Wesleyan University Press: Middletown, CT, USA, 2011. [Google Scholar]
- Gymrek, M.; McGuire, A.L.; Golan, D.; Halperin, E.; Erlich, Y. Identifying Personal Genomes by Surname Inference. Science 2013, 339, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Loreille, O.M.; Diegoli, T.M.; Irwin, J.A.; Coble, M.D.; Parsons, T.J. High Efficiency DNA Extraction from Bone by Total Demineralization. Forensic Sci. Int. Genet. 2007, 1, 191–195. [Google Scholar] [CrossRef]
- Gorden, E.M.; Sturk-Andreaggi, K.; Marshall, C. Repair of DNA Damage Caused by Cytosine Deamination in Mitochondrial DNA of Forensic Case Samples. Forensic Sci. Int. Genet. 2018, 34, 257–264. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Short Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Picard. Available online: https://broadinstitute.github.io/picard/ (accessed on 23 February 2019).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- The Snipper App Suite, version 2.5. Available online: http://mathgene.usc.es/snipper/index.php (accessed on 23 February 2019).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A.; Mayrose, I. Clumpak: A Program for Identifying Clustering Modes and Packaging Population Structure Inferences Across K. Mol. Ecol. Resour. 2015, 15, 1179–1191. [Google Scholar] [CrossRef] [PubMed]
- Cann, H.M.; De Toma, C.; Cazes, L.; Legrand, M.; Morel, V.; Piouffre, L.; Bodmer, J.; Bodmer, W.F.; Bonne-Tamir, B.; Cambon-Thomsen, A. A Human Genome Diversity Cell Line Panel. Science 2002, 296, 261–262. [Google Scholar] [CrossRef] [PubMed]
- 1000 Genomes Project Consortium. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A. The Simons Genome Diversity Project: 300 Genomes from 142 Diverse Populations. Nature 2016, 538, 201. [Google Scholar] [CrossRef] [PubMed]
- Sturk, K.A.; Coble, M.D.; Barritt, S.M.; Irwin, J.A. Evaluation of Modified Yfiler Amplification Strategy for Compromised Samples. Croat. Med. J. 2009, 50, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Y-DNA Haplogroup Predictor–NevGen. Available online: www.nevgen.org (accessed on 15 January 2019).
- England, R.; Curnow, N.; Liu, A.; Stacey, J.; Harbison, S. Massively Parallel Sequencing of Identifiler and PowerPlex® Y Amplified Forensic Samples. Forensic Sci. Int. Genet. Suppl. Ser. 2015, 5, e210–e212. [Google Scholar] [CrossRef]
- Warshauer, D.H.; Lin, D.; Hari, K.; Jain, R.; Davis, C.; Larue, B.; King, J.L.; Budowle, B. STRait Razor: A Length-Based Forensic STR Allele-Calling Tool for use with Second Generation Sequencing Data. Forensic. Sci. Int. Genet. 2013, 7, 409–417. [Google Scholar] [CrossRef]
- Myres, N.M.; Rootsi, S.; Lin, A.A.; Järve, M.; King, R.J.; Kutuev, I.; Cabrera, V.M.; Khusnutdinova, E.K.; Pshenichnov, A.; Yunusbayev, B. A Major Y-Chromosome Haplogroup R1b Holocene Era Founder Effect in Central and Western Europe. Eur. J. Hum. Genet. 2011, 19, 95. [Google Scholar] [CrossRef]
- R R1b ALL Subclades. Available online: https://www.familytreedna.com/groups/r-1b/about/background (accessed on 15 May 2019).
- King, T.E.; Jobling, M.A. Founders, Drift, and Infidelity: The Relationship between Y Chromosome Diversity and Patrilineal Surnames. Mol. Biol. Evol. 2009, 26, 1093–1102. [Google Scholar] [CrossRef]
- Hale, C.R. Charles R. Hale Collection [of Cemetery Inscriptions and Newspaper Notices of Marriages and Deaths]; Connecticut State Library: Hartford, CT, USA, 1933; p. 34. [Google Scholar]
- Greytak, E.M.; Moore, C.; Armentrout, S.L. Genetic Genealogy for Cold Case and Active Investigations. Forensic Sci. Int. 2019, 299, 103–113. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Fortes, G.G.; Balaresque, P.; Thomas, M.G.; Balding, D.; Delser, P.M.; Neumann, R.; Parson, W.; Knapp, M.; Walsh, S. Identification of the Remains of King Richard III. Nat. Commun. 2014, 5, 5631. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.L.; Wadhams, M.J.; Roby, R.K.; Holland, M.M.; Weedn, V.W.; Parsons, T.J. Mitochondrial DNA Sequence Heteroplasmy in the Grand Duke of Russia Georgij Romanov Establishes the Authenticity of the Remains of Tsar Nicholas II. Nat. Genet. 1996, 12, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Gill, P.; Ivanov, P.L.; Kimpton, C.; Piercy, R.; Benson, N.; Tully, G.; Evett, I.; Hagelberg, E.; Sullivan, K. Identification of the Remains of the Romanov Family by DNA Analysis. Nat. Genet. 1994, 6, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Coble, M.D.; Loreille, O.M.; Wadhams, M.J.; Edson, S.M.; Maynard, K.; Meyer, C.E.; Niederstatter, H.; Berger, C.; Berger, B.; Falsetti, A.B.; et al. Mystery Solved: The Identification of the Two Missing Romanov Children using DNA Analysis. PLoS ONE 2009, 4, e4838. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Autosomal DNA Concentration (99 bp target) (ng/µL) | Y-Chromosomal DNA Concentration (133 bp target) (ng/µL) | Autosomal: Y-Chromosomal DNA Ratio |
|---|---|---|---|
| JB55-1 | 0.2009 | 0.0871 | 2.3072 |
| RB-1 | 0 | 0 | 0 |
| JB55-2 | 0.1735 | 0.0802 | 2.1624 |
| RB-2 | 0 | 0 | 0 |
| Locus | JB55-1 Allele(s) | JB55-2 Allele(s) |
|---|---|---|
| DYS19 | 15 | 15 |
| DYS385 | 11,13 | 11,13 |
| DYS389I | 13 | 13 |
| DYS389II | 29 | 29 |
| DYS390 | 23 | 23 |
| DYS391 | 11 | 11 |
| DYS392 | 13 | 13 |
| DYS393 | 13 | 13 |
| DYS437 | 15 | 15 |
| DYS438 | 12 | No Data |
| DYS439 | 12 | 12 |
| DYS448 | 19 | 19 |
| DYS456 | 15 | 15 |
| DYS458 | 17 | 17 |
| DYS635 | 23 | 23 |
| Y GATA H4 | 12 | 12 |
| JB55-1 | JB55-2 | ||||
|---|---|---|---|---|---|
| Allele | Read Count | % of Total Reads | Allele | Read Count | % of Total Reads |
| 12 | 12,343 | 96.3% | 12 | 8 | 88.9% |
| 11 | 386 | 3.0% | 13 | 1 | 11.1% |
| Other | 87 | 0.7% | Other | 0 | 0% |
| Total | 12,816 | 100% | Total | 9 | 100% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daniels-Higginbotham, J.; Gorden, E.M.; Farmer, S.K.; Spatola, B.; Damann, F.; Bellantoni, N.; Gagnon, K.S.; de la Puente, M.; Xavier, C.; Walsh, S.; et al. DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes 2019, 10, 636. https://doi.org/10.3390/genes10090636
Daniels-Higginbotham J, Gorden EM, Farmer SK, Spatola B, Damann F, Bellantoni N, Gagnon KS, de la Puente M, Xavier C, Walsh S, et al. DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes. 2019; 10(9):636. https://doi.org/10.3390/genes10090636
Chicago/Turabian StyleDaniels-Higginbotham, Jennifer, Erin M. Gorden, Stephanie K. Farmer, Brian Spatola, Franklin Damann, Nicholas Bellantoni, Katie S. Gagnon, Maria de la Puente, Catarina Xavier, Susan Walsh, and et al. 2019. "DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut" Genes 10, no. 9: 636. https://doi.org/10.3390/genes10090636
APA StyleDaniels-Higginbotham, J., Gorden, E. M., Farmer, S. K., Spatola, B., Damann, F., Bellantoni, N., Gagnon, K. S., de la Puente, M., Xavier, C., Walsh, S., Parson, W., McMahon, T. P., & Marshall, C. (2019). DNA Testing Reveals the Putative Identity of JB55, a 19th Century Vampire Buried in Griswold, Connecticut. Genes, 10(9), 636. https://doi.org/10.3390/genes10090636