Integration of Transcriptomes, Small RNAs, and Degradome Sequencing to Identify Putative miRNAs and Their Targets Related to Eu-Rubber Biosynthesis in Eucommia ulmoides

Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Growth Conditions
2.2. Total RNA Preparation
2.3. Transcriptome Sequencing and De Novo Assembly Analysis
2.4. sRNA Sequencing and miRNAs Identification
2.5. Degradome Sequencing and Target Identification and Prediction
2.6. Functional Annotation of Targets
2.7. RLM-5′RACE to Verify the Identified and Predicted Targets
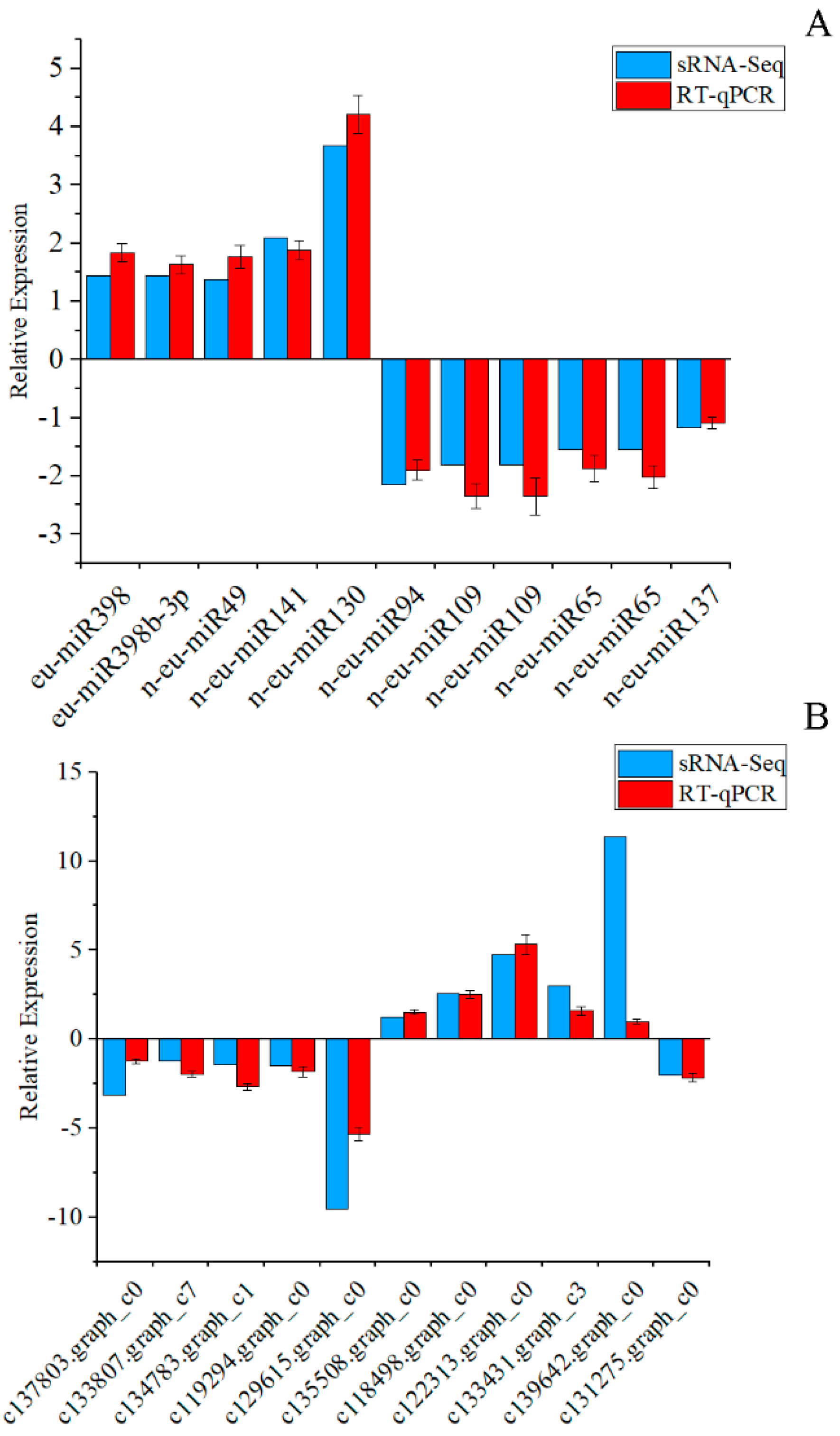
2.8. Validation of miRNAs’ and Their Targets’ Expression by RT-qPCR
3. Results
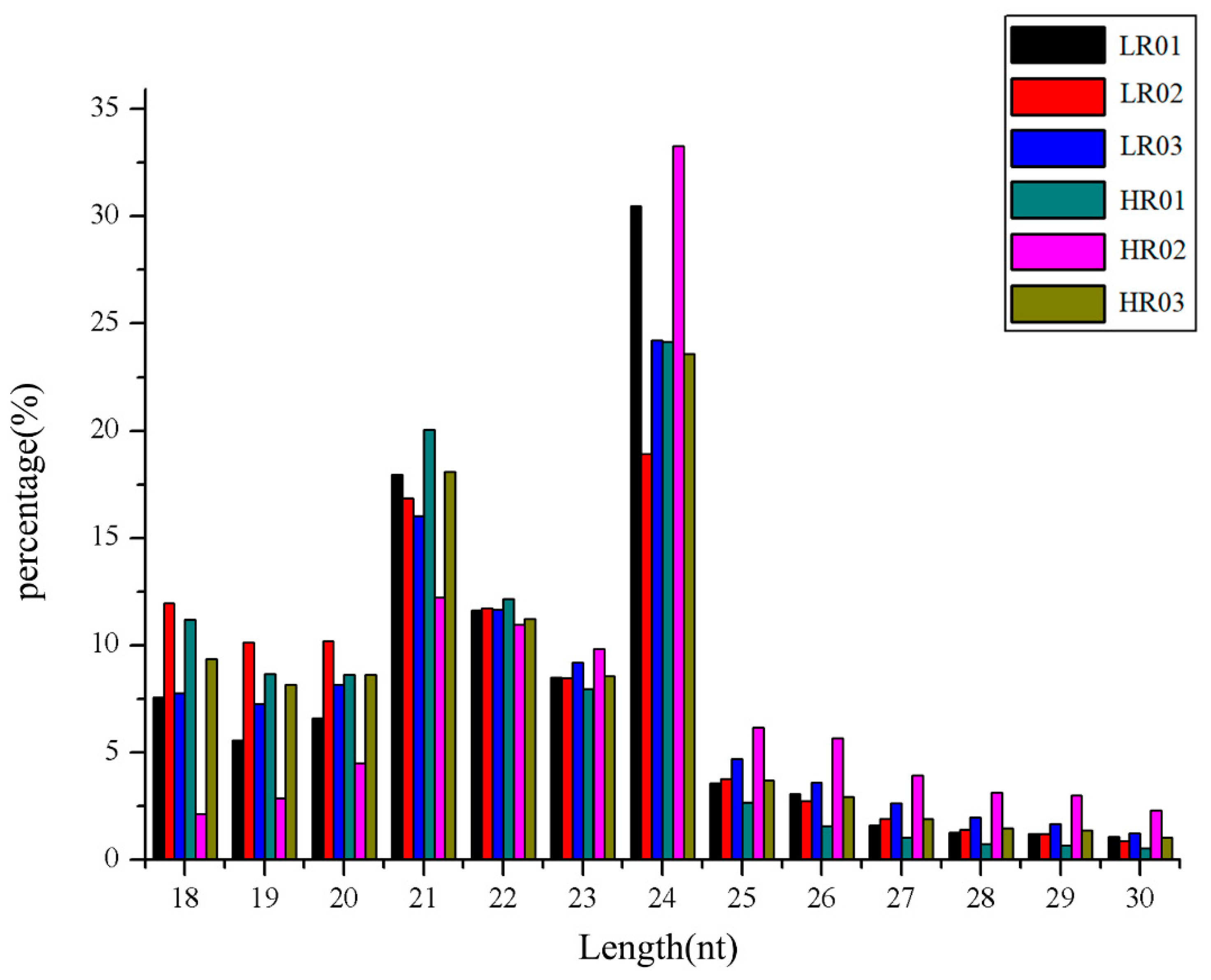
3.1. Overview of mRNAs and sRNAs Sequencing Data
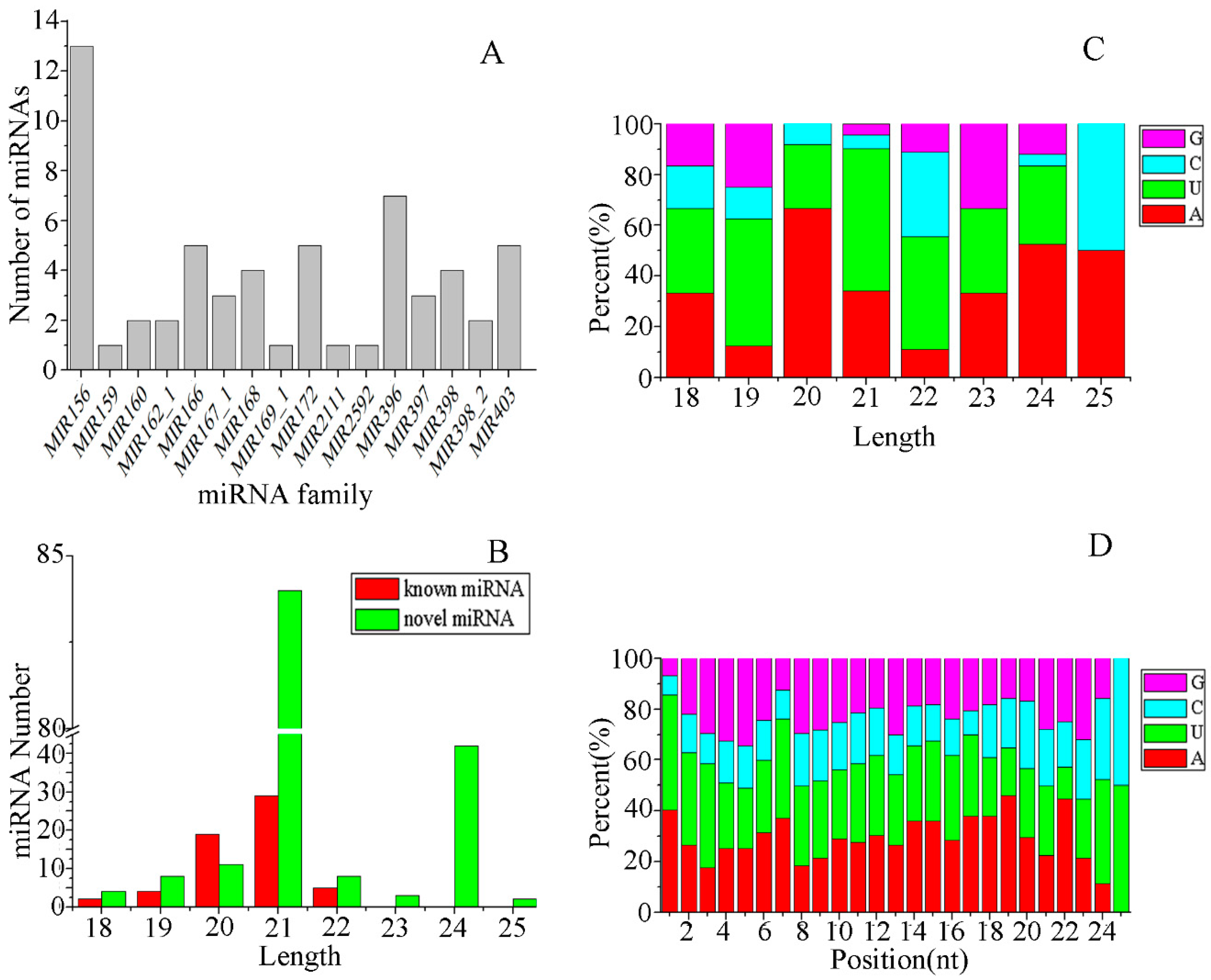
3.2. Identification of miRNAs in E. ulmoides
3.3. Eu-Rubber Biosynthesis-Related miRNAs
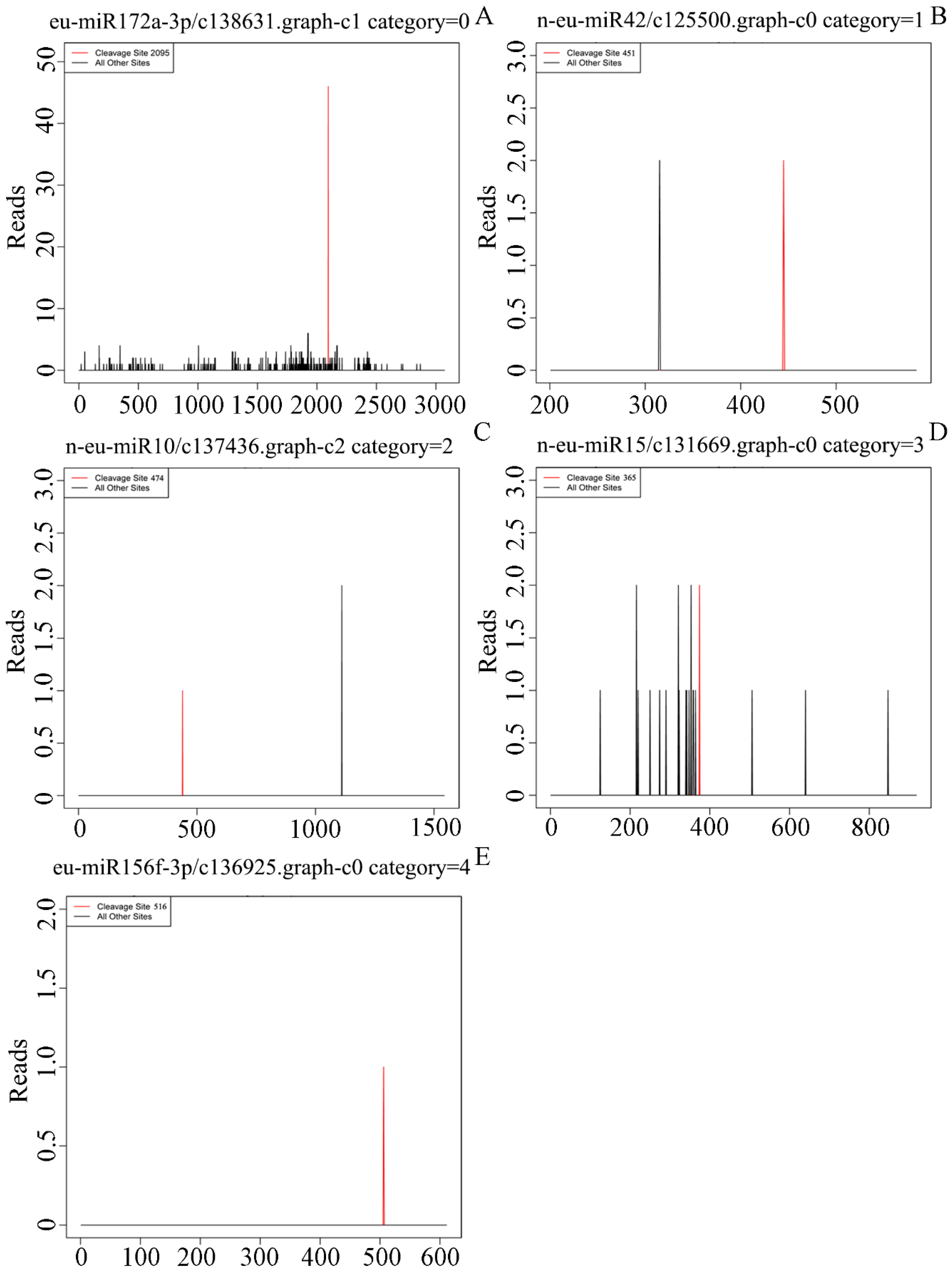
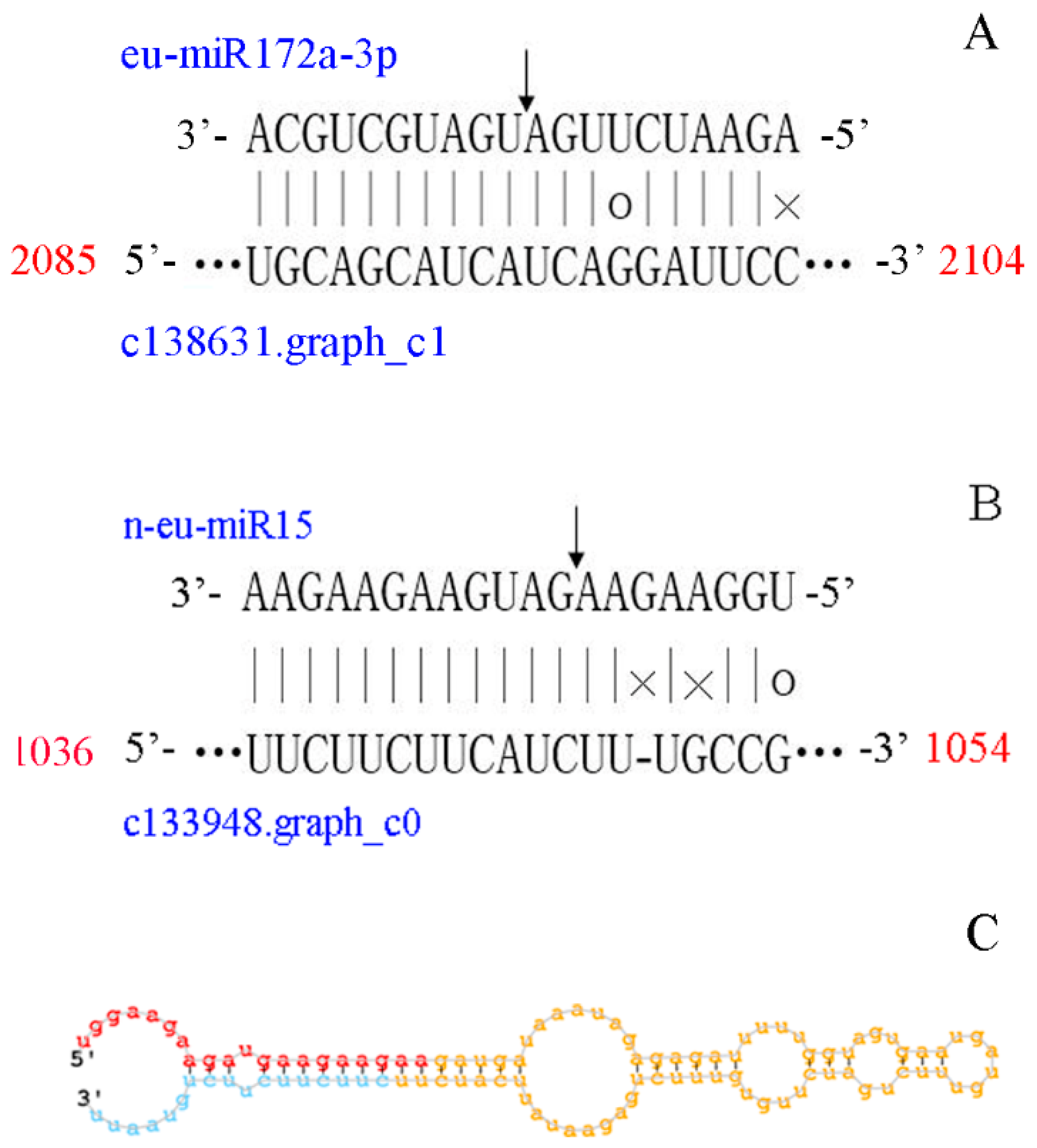
3.4. A Target Prediction, Identification and Validation
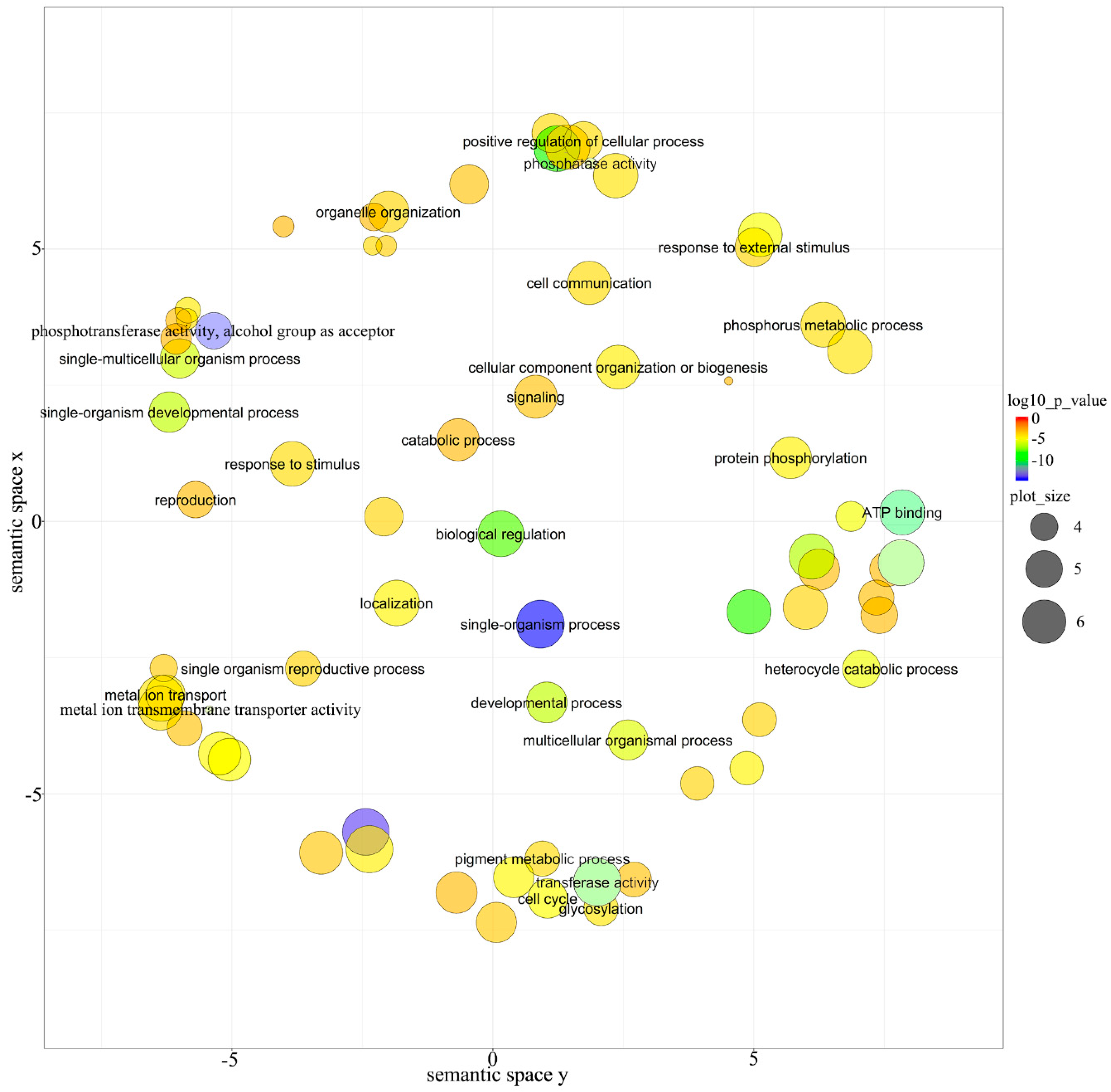
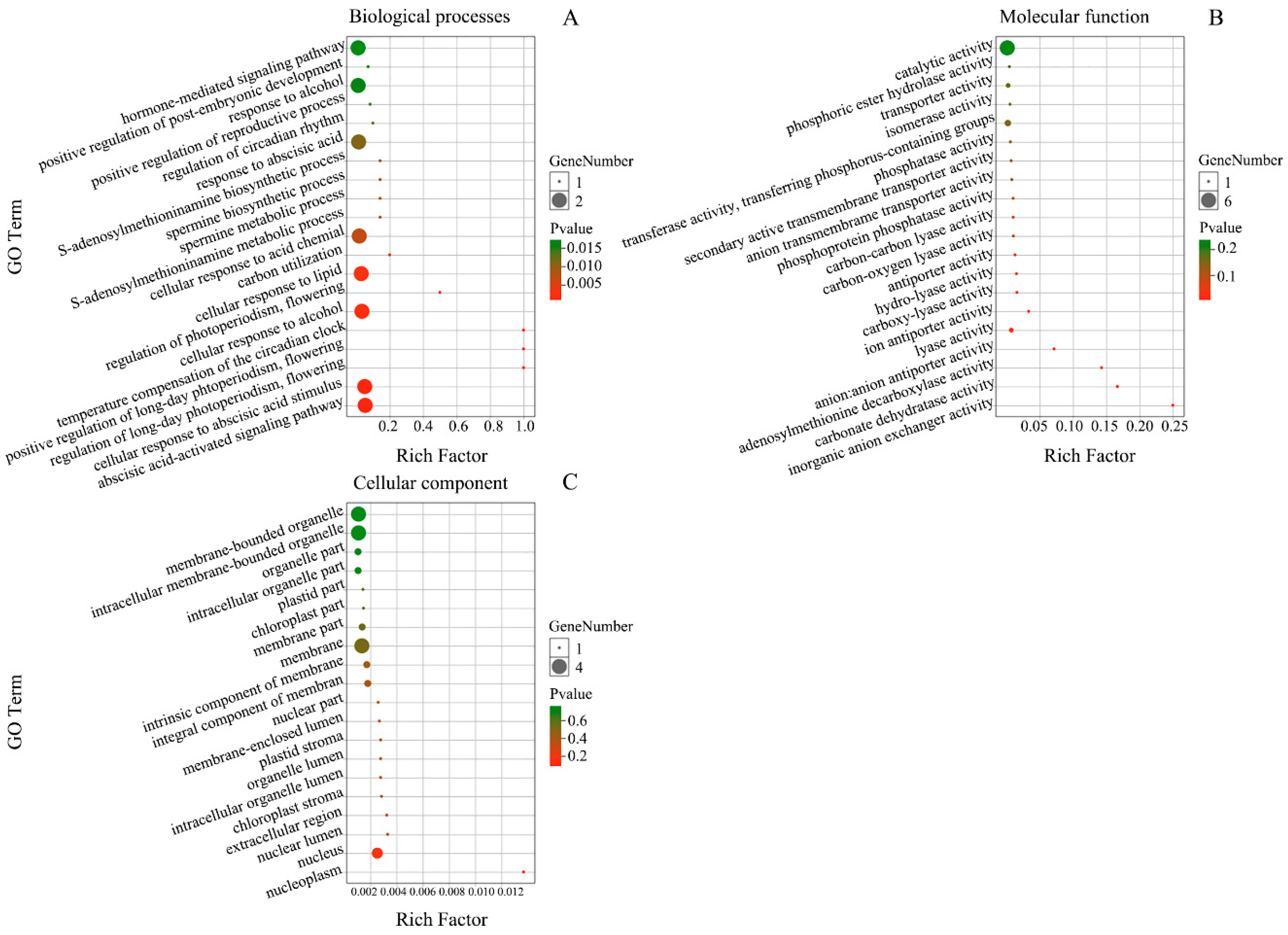
3.5. Function Analysis of Targets for miRNAs in E. ulmoides
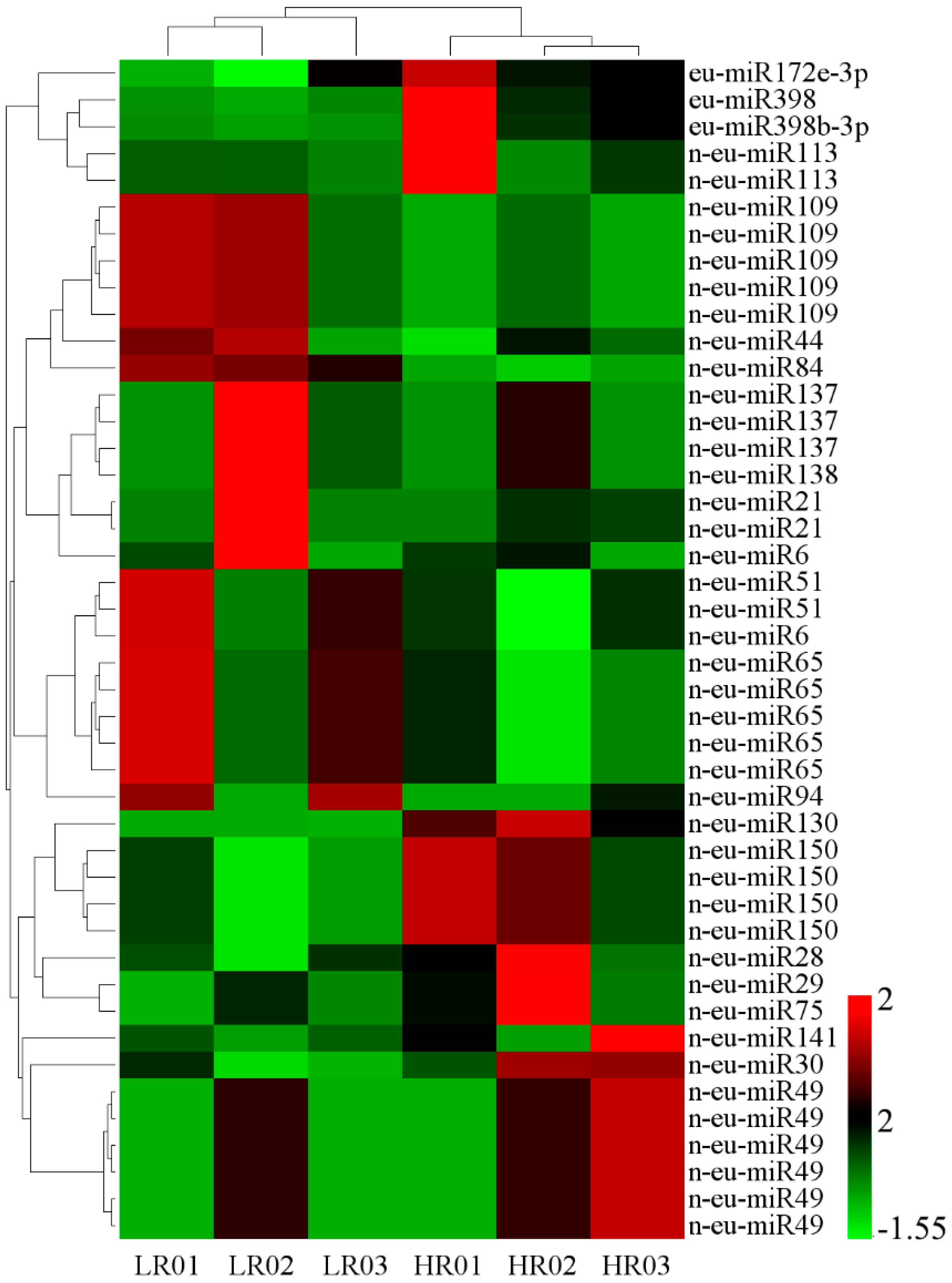
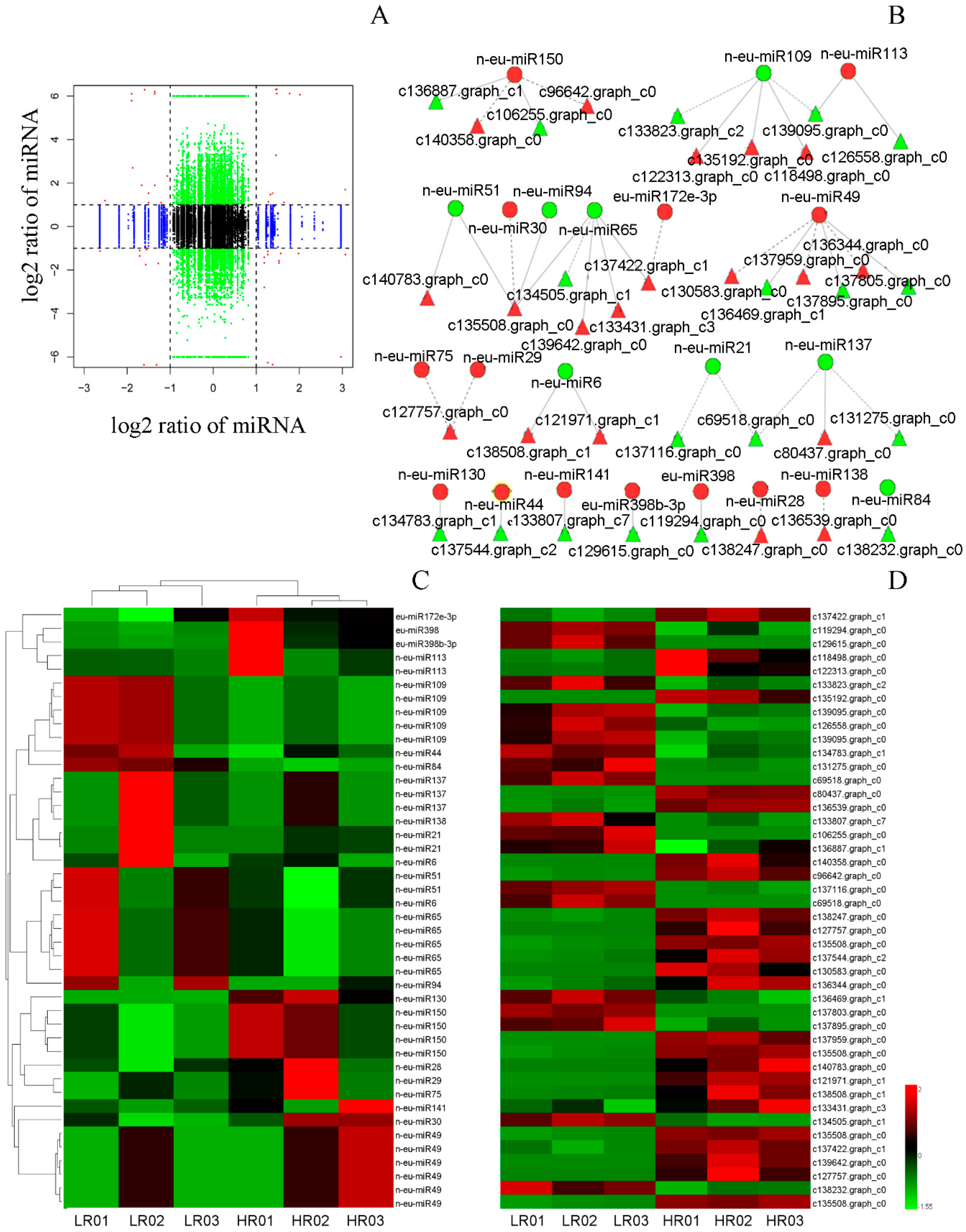
3.6. Correlation Analysis of miRNAs and Their Targets’ Expression Profiles
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jover-Gil, S.; Candela, H.; Ponce, M.R. Plant micrornas and development. Int. J. Dev. Biol. 2005, 49, 733. [Google Scholar] [CrossRef] [PubMed]
- Voinnet, O. Origin, biogenesis, and activity of plant micrornas. Cell 2009, 136, 669–687. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Wang, Y.; Zhai, L.; Xu, Y.; Wang, L.; Zhu, X.; Gong, Y.; Yu, R.; Limera, C.; Liu, L. Genome-wide identification and characterization of cadmium-responsive micrornas and their target genes in radish (Raphanus sativus L.) roots. J. Exp. Bot. 2013, 64, 4271–4287. [Google Scholar] [CrossRef] [PubMed]
- Mangrauthia, S.K.; Bhogireddy, S.; Agarwal, S.; Prasanth, V.V.; Voleti, S.R.; Neelamraju, S.; Subrahmanyam, D. Genome-wide changes in microrna expression during short and prolonged heat stress and recovery in contrasting rice cultivars. J. Exp. Bot. 2017, 68, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Quan, S.; Xu, H.; Ma, L.; Niu, J. Identification and expression of mirnas related to female flower induction in walnut (Juglans regia L.). Molecules 2018, 23, 1202. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Cao, Z. Genome-wide expression profiling of micrornas in poplar upon infection with the foliar rust fungus melampsora larici-populina. BMC Genom. 2015, 16, 696. [Google Scholar] [CrossRef] [PubMed]
- Davuluri, G.; Van Tuinen, A.; Fraser, P.D.; Manfredonia, A.; Newman, R.; Burgess, D.; Brummell, D.; King, S.; Palys, J.; Uhlig, J. Fruit-specific rnai-mediated suppression of det1 enhances carotenoid and flavonoid content in tomatoes. Nat. Biotechnol. 2005, 23, 890. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.Y.; Felippes, F.F.; Liu, C.J.; Weigel, D.; Wang, J.W. Negative regulation of anthocyanin biosynthesis in arabidopsis by a mir156-targeted spl transcription factor. Plant Cell 2011, 23, 1512–1522. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.W.-K.; Zhang, C.; Miller, M.; Palmer, G.; Whiteley, M.; Tholl, D.; Chen, Z.J. Cis- and trans-regulation of mir163 and target genes confers natural variation of secondary metabolites in two arabidopsis species and their allopolyploids. Plant Cell 2011, 23, 1729–1740. [Google Scholar] [CrossRef]
- Wang, L.; Du, H.; Wuyun, T.N. Genome-wide identification of micrornas and their targets in the leaves and fruits of eucommia ulmoides using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef]
- Gébelin, V.; Leclercq, J.; Kuswanhadi; Argout, X.; Chaidamsari, T.; Hu, S.; Tang, C.; Sarah, G.; Yang, M.; Montoro, P. The small RNA profile in latex from hevea brasiliensis trees is affected by tapping panel dryness. Tree Physiol. 2013, 33, 1084. [Google Scholar]
- Pramoolkit, P.; Lertpanyasampatha, M.; Viboonjun, U.; Kongsawadworakul, P.; Chrestin, H.; Narangajavana, J. Involvement of ethylene-responsive micrornas and their targets in increased latex yield in the rubber tree in response to ethylene treatment. Plant Physiol. Biochem. 2014, 84, 203–212. [Google Scholar] [CrossRef]
- Wuyun, T.N.; Wang, L.; Liu, H.; Wang, X.; Zhang, L.; Bennetzen, J.L.; Li, T.; Yang, L.; Liu, P.; Du, L. The hardy rubber tree genome provides insights into the evolution of polyisoprene biosynthesis. Mol. Plant 2018, 11, 429. [Google Scholar] [CrossRef]
- Ae, P.S.; Myung-Sook, C.; Un Ju, J.; Myung-Joo, K.; Ju, K.D.; Hae-Mo, P.; Bok, P.Y.; Mi-Kyung, L. Eucommia ulmoides oliver leaf extract increases endogenous antioxidant activity in type 2 diabetic mice. J. Med. Food 2006, 9, 474–479. [Google Scholar]
- Takeno, S.; Bamba, T.; Nakazawa, Y.; Fukusaki, E.; Okazawa, A.; Kobayashi, A. Quantification of trans-1,4-polyisoprene in eucommia ulmoides by fourier transform infrared spectroscopy and pyrolysis-gas chromatography/mass spectrometry. J. Biosci. Bioeng. 2008, 105, 355–359. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Takeda, T.; Suzuki, N.; Hayashi, T.; Harada, Y.; Bamba, T.; Kobayashi, A. Histochemical study of trans-polyisoprene accumulation by spectral;confocal laser scanning microscopy and a specific dye showing;fluorescence solvatochromism in the rubber-producing plant, eucommia; ulmoides oliver. Planta 2013, 238, 549–560. [Google Scholar] [CrossRef]
- Lin, T.; Xu, X.; Ruan, J.; Liu, S.; Wu, S.; Shao, X.; Wang, X.; Gan, L.; Qin, B.; Yang, Y.; et al. Genome analysis of taraxacum kok-saghyz rodin provides new insights into rubber biosynthesis. Natl. Sci. Rev. 2018, 5, 78–87. [Google Scholar] [CrossRef]
- Mooibroek, H.; Cornish, K. Alternative sources of natural rubber. Appl. Microbiol. Biotechnol. 2000, 53, 355–365. [Google Scholar] [CrossRef]
- Lieberei, R. South american leaf blight of the rubber tree (hevea spp.): New steps in plant domestication using physiological features and molecular markers. Ann. Bot. 2007, 100, 1125–1142. [Google Scholar] [CrossRef]
- Nair, K.P.P. The Agronomy and Economy of Important Tree Crops of the Developing World; Elsevier: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Ren, C.; Harada, Y.; Bamba, T.; Nakazawa, Y.; Gyokusen, K. Overexpression of an isopentenyl diphosphate isomerase gene to enhance trans -polyisoprene production in eucommia ulmoides oliver. BMC Biotechnol. 2012, 12, 78. [Google Scholar]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. Mirdeep2 accurately identifies known and hundreds of novel microrna genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, L.; Wang, J.; Gu, P.; Chen, M. Mtide: An integrated tool for the identification of mirna–target interaction in plants. Bioinformatics 2015, 31, 290–291. [Google Scholar] [CrossRef]
- Addo-Quaye, C.; Miller, W.A.; Axtel, M.J. Cleaveland: A pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics 2009, 25, 130–131. [Google Scholar] [CrossRef]
- Charles, A.-Q.; Eshoo, T.W.; Bartel, D.P.; Axtel, M.J. Endogenous sirna and mirna targets identified by sequencing of the arabidopsis degradome. Curr. Biol. 2008, 18, 758–762. [Google Scholar]
- Dai, X.; Zhao, P.X. Psrnatarget: A plant small RNA target analysis server. Nucleic Acids Res. 2011, 39, W155. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, X.-J. Goeast: A web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008, 36, W358–W363. [Google Scholar] [CrossRef]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. Revigo summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. Kaas: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. Go: Termfinder—open source software for accessing gene ontology information and finding significantly enriched gene ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef]
- Liu, X.; Chen, M.; Zhou, X.; Cao, Z. Identification of novel mirnas and their target genes from populus szechuanica infected with melampsora larici-populina. Mol. Biol. Rep. 2019, 46, 3083–3092. [Google Scholar] [CrossRef]
- Ye, J.; Jin, C.-F.; Li, N.; Liu, M.-H.; Fei, Z.-X.; Dong, L.-Z.; Li, L.; Li, Z.-Q. Selection of suitable reference genes for qRT-PCR normalisation under different experimental conditions in eucommia ulmoides oliv. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Huang, H. Development of ssr molecular markers based on transcriptome sequencing of eucommia ulmoides. Sci. Silvae Sin. 2013, 49, 176–181. [Google Scholar]
- Nobuaki, S.; Hirotaka, U.; Takashi, N.; Yukio, M.; Atsushi, Y.; Masahira, H.; Naotake, O.; Takeshi, B.; Ei-Ichiro, F.; Akio, K. Construction and analysis of est libraries of the trans-polyisoprene producing plant, eucommia ulmoides oliver. Planta 2012, 236, 1405–1417. [Google Scholar]
- Qi, W.A.; Qi, H.L.; Shao, A.J.; Hong, C.G.; Min, C.; Hui, T.C. Genetic diversity of eucommia ulmoides by rapd analysis. China J. Chin. Mater. Med. 2006, 31, 1583. [Google Scholar]
- Li, Y.; Han, C.; Wang, J.; Xiao, W.; Wang, Z.; Zhang, J.; Yang, Y.; Zhang, S.; Ai, C. Investigation into the mechanism of eucommia ulmoides oliv. Based on a systems pharmacology approach. J. Ethnopharmacol. 2014, 151, 452–460. [Google Scholar] [CrossRef]
- Yang, Z.M.; Chen, J. A potential role of micrornas in plant response to metal toxicity. Met. Integr. Biometal Sci. 2013, 5, 1184–1190. [Google Scholar] [CrossRef]
- Xie, F.; Wang, Q.; Sun, R.; Zhang, B. Deep sequencing reveals important roles of micrornas in response to drought and salinity stress in cotton. J. Exp. Bot. 2015, 66, 789–804. [Google Scholar] [CrossRef]
- Long, J.M.; Liu, Z.; Wu, X.M.; Fang, Y.N.; Jia, H.H.; Xie, Z.Z.; Deng, X.X.; Guo, W.W. Genome-scale mrna and small RNA transcriptomic insights into initiation of citrus apomixis. J. Exp. Bot. 2016, 67, 5743–5756. [Google Scholar] [CrossRef]
- Ming, D.; Meihua, H.; Fen, L.; Renyuan, Q.; Xiaojun, L.; Ezhen, Z.; Maokang, H.; Zhenyong, H.; Quanguang, H.E. Identification of ethylene responsive mirnas and their targets from newly harvested banana fruits using high-throughput sequencing. J. Agric. Food Chem. 2018, 66, 10628–10639. [Google Scholar]
- Yang, J.; Liu, X.; Xu, B.; Zhao, N.; Yang, X.; Zhang, M. Identification of mirnas and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of brassica juncea. BMC Genom. 2013, 14, 9. [Google Scholar] [CrossRef]
- Baksa, I.; Nagy, T.; Barta, E.; Havelda, Z.; Várallyay, É.; Silhavy, D.; Burgyán, J.; Szittya, G. Identification of nicotiana benthamiana micrornas and their targets using high throughput sequencing and degradome analysis. BMC Genom. 2015, 16, 1025. [Google Scholar] [CrossRef]
- Fabio, F.; George, C. Plant phase transitions make a splash. Cell 2009, 138, 625–627. [Google Scholar]
- Wu, G.; Poethig, R.S. Temporal regulation of shoot development in arabidopsis thaliana by mir156 and its target spl3. Development 2006, 133, 3539–3547. [Google Scholar] [CrossRef]
- Han, X.; Yin, H.; Song, X.; Zhang, Y.; Liu, M.; Sang, J.; Jiang, J.; Li, J.; Zhuo, R. Integration of small rnas, degradome and transcriptome sequencing in hyperaccumulator sedum alfredii uncovers a complex regulatory network and provides insights into cadmium phytoremediation. Plant Biotechnol. J. 2016, 14, 1470–1483. [Google Scholar] [CrossRef]
- Xuemei, C. A microrna as a translational repressor of apetala2 in arabidopsis flower development. Science 2004, 303, 2022–2025. [Google Scholar]
- Holt, D.B.; Gupta, V.; Meyer, D.; Abel, N.B.; Andersen, S.U.; Stougaard, J.; Markmann, K. Micro RNA 172 (mir172) signals epidermal infection and is expressed in cells primed for bacterial invasion in lotus japonicus roots and nodules. New Phytol. 2015, 208, 241–256. [Google Scholar] [CrossRef]
- Nova-Franco, B.; Íñiguez, L.P.; Valdés-López, O.; Alvarado-Affantranger, X.; Leija, A.; Fuentes, S.I.; Ramírez, M.; Paul, S.; Reyes, J.L.; Girard, L.; et al. The micro-rna72c-apetala2-1 node as a key regulator of the common bean-rhizobium etli nitrogen fixation symbiosis. Plant Physiol. 2015, 168, 273–291. [Google Scholar] [CrossRef]
- Youning, W.; Lixiang, W.; Yanmin, Z.; Liang, C.; Zhaoming, C.; Senlei, Z.; Fang, Z.; Yinping, T.; Qiong, J.; Ferguson, B.J. Soybean mir172c targets the repressive ap2 transcription factor nnc1 to activate enod40 expression and regulate nodule initiation. Plant Cell 2014, 26, 4782–4801. [Google Scholar]
- Yan, Z.; Hossain, M.S.; Wang, J.; Valdes-Lopez, O.; Liang, Y.; Libault, M.; Qiu, L.; Stacey, G. Mir172 regulates soybean nodulation. Mol. Plant Microbe Interact. 2013, 26, 1371–1377. [Google Scholar] [CrossRef]
- Wu, G.; Park, M.Y.; Conway, S.R.; Wang, J.W.; Weigel, D.; Poethig, R.S. The sequential action of mir156 and mir172 regulates developmental timing in arabidopsis. Cell 2009, 138, 750–759. [Google Scholar] [CrossRef]
- Sahito, Z.A.; Wang, L.; Sun, Z.; Yan, Q.; Zhang, X.; Jiang, Q.; Ullah, I.; Tong, Y.; Li, X. The mir172c-nnc1 module modulates root plastic development in response to salt in soybean. BMC Plant Biol. 2017, 17, 229. [Google Scholar] [CrossRef]
- Jagadeeswaran, G.; Saini, A.; Sunkar, R. Biotic and abiotic stress down-regulate mir398 expression in arabidopsis. Planta 2009, 229, 1009–1014. [Google Scholar] [CrossRef]
- Ramanjulu, S. Posttranscriptional induction of two cu/zn superoxide dismutase genes in arabidopsis is mediated by downregulation of mir398 and important for oxidative stress tolerance. Plant Cell 2006, 8. [Google Scholar]
- De, l.R.C.; Covarrubias, A.A.; Reyes, J.L. A dicistronic precursor encoding mir398 and the legume-specific mir2119 co-regulates csd1 and adh1 mrnas in response to water deficit. Plant Cell Environ. 2018, 42, 133–144. [Google Scholar]
- Yin, T.; Cao, X.; Qian, M.; Li, C.; Chen, X.; Zhou, M.; Jiang, J. Molecular cloning and functional analysis of an organ-specific expressing gene coding for farnesyl diphosphate synthase from michelia chapensis dandy. Acta Physiol. Plant. 2011, 33, 137–144. [Google Scholar] [CrossRef]
- Wei, R.; Qiu, D.; Wilson, I.W.; Zhao, H.; Lu, S.; Miao, J.; Feng, S.; Bai, L.; Wu, Q.; Tu, D. Identification of novel and conserved micrornas in panax notoginseng roots by high-throughput sequencing. BMC Genom. 2015, 16, 835. [Google Scholar] [CrossRef]
- Brodersen, P.; Sakvarelidzeachard, L.; Bruunrasmussen, M.; Dunoyer, P.; Yamamoto, Y.Y.; Sieburth, L.; Voinnet, O. Widespread translational inhibition by plant mirnas and sirnas. Science 2008, 320, 1185–1190. [Google Scholar] [CrossRef]
- Pei, H.; Ma, N.; Chen, J.; Zheng, Y.; Tian, J.; Li, J.; Zhang, S.; Fei, Z.; Gao, J. Integrative analysis of mirna and mrna profiles in response to ethylene in rose petals during flower opening. PLoS ONE 2013, 8, e64290. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target ID | miRNAs | Target annotation |
|---|---|---|
| c146902.graph_c0 | n-eu-miR39 | geranylgeranyl diphosphate reductase |
| c128958.graph_c4 | n-eu-miR85 | ditrans,polycis-polyprenyl diphosphate synthase |
| c134980.graph_c0 | n-eu-miR5 | ditrans,polycis-polyprenyl diphosphate synthase |
| c130182.graph_c1 | n-eu-miR45 | 1-deoxy-D-xylulose-5-phosphate reductoisomerase |
| c138767.graph_c0 | n-eu-miR65 | 1-deoxy-D-xylulose-5-phosphate synthase |
| c132996.graph_c3 | eu-miR156f-5p | prenyl protein peptidase |
| c148011.graph_c0 | n-eu-miR76 | geranylgeranyl diphosphate synthase, type II |
| c130198.graph_c0 | eu-miR156f-3p | farnesyl diphosphate synthase |
| c139976.graph_c0 | n-eu-miR42 | geranylgeranyl diphosphate synthase, type II |
| c124585.graph_c0 | n-eu-miR43 | hydroxymethylglutaryl-CoA reductase (NADPH) |
| c137756.graph_c2 | n-eu-miR91 | phosphomevalonate kinase |
| c96211.graph_c0 | n-eu-miR42 | endopeptidase |
| c136111.graph_c2 | n-eu-miR42; n-eu-miR152 | farnesyl diphosphate synthase |
| c122494.graph_c0 | n-eu-miR1; n-eu-miR8 | acetyl-CoA C-acetyltransferase |
| c138663.graph_c0 | eu-miR396b; n-eu-miR60 | diphosphomevalonate decarboxylase |
| c126798.graph_c1 | n-eu-miR85; n-eu-miR47 | prenylcysteine α-carboxyl methylesterase |
| c133948.graph_c0 | eu-miR396c-3p; n-eu-miR15; n-eu-miR109 | geranylgeranyl diphosphate synthase, type II |
| c136533.graph_c5 | eu-miR156b; n-eu-miR66; n-eu-miR101; n-eu-miR147 | farnesyl diphosphate synthase |
| c121732.graph_c0 | eu-miR2111a-5p; n-eu-miR1; n-eu-miR8; n-eu-miR66 | 1-deoxy-D-xylulose-5-phosphate synthase |
| c139224.graph_c1 | eu-miR156b; n-eu-miR43; n-eu-miR59; n-eu-miR108 | prenylcysteine oxidase / farnesylcysteine lyase |
| c136892.graph_c1 | n-eu-miR1; n-eu-miR4; n-eu-miR94; n-eu-miR95; n-eu-miR142; n-eu-miR152 | all-trans-nonaprenyl-diphosphate synthase |
| c137351.graph_c2 | n-eu-miR1; n-eu-miR7; n-eu-miR8; n-eu-miR33; n-eu-miR43; n-eu-miR45; n-eu-miR76; n-eu-miR78; n-eu-miR152 | isopentenyl-diphosphate delta-isomerase |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, J.; Han, W.; Fan, R.; Liu, M.; Li, L.; Jia, X. Integration of Transcriptomes, Small RNAs, and Degradome Sequencing to Identify Putative miRNAs and Their Targets Related to Eu-Rubber Biosynthesis in Eucommia ulmoides. Genes 2019, 10, 623. https://doi.org/10.3390/genes10080623
Ye J, Han W, Fan R, Liu M, Li L, Jia X. Integration of Transcriptomes, Small RNAs, and Degradome Sequencing to Identify Putative miRNAs and Their Targets Related to Eu-Rubber Biosynthesis in Eucommia ulmoides. Genes. 2019; 10(8):623. https://doi.org/10.3390/genes10080623
Chicago/Turabian StyleYe, Jing, Wenjing Han, Ruisheng Fan, Minhao Liu, Long Li, and Xiaoming Jia. 2019. "Integration of Transcriptomes, Small RNAs, and Degradome Sequencing to Identify Putative miRNAs and Their Targets Related to Eu-Rubber Biosynthesis in Eucommia ulmoides" Genes 10, no. 8: 623. https://doi.org/10.3390/genes10080623
APA StyleYe, J., Han, W., Fan, R., Liu, M., Li, L., & Jia, X. (2019). Integration of Transcriptomes, Small RNAs, and Degradome Sequencing to Identify Putative miRNAs and Their Targets Related to Eu-Rubber Biosynthesis in Eucommia ulmoides. Genes, 10(8), 623. https://doi.org/10.3390/genes10080623